I did some exercises to find the ideal strength of the potential energy function (Fe) which I’m using now. And I have some question.
Part 1:
I used my first file to change the length (Lattice constant) of Fe in the z direction. Then draw the figure about distance z and pressure Y(Z-Py). When the derivative is 0, as the ideal strength. (fig and script flow the text)
Q1: Am I using the right method to find the ideal strength of Fe? Because when I use ovito to check my dump file. I saw that distance between the atoms in each layer is very large. There is my fig and script: I use the .sh file to continuously change the parameters (like hogehogehoge etc)
Z-Py
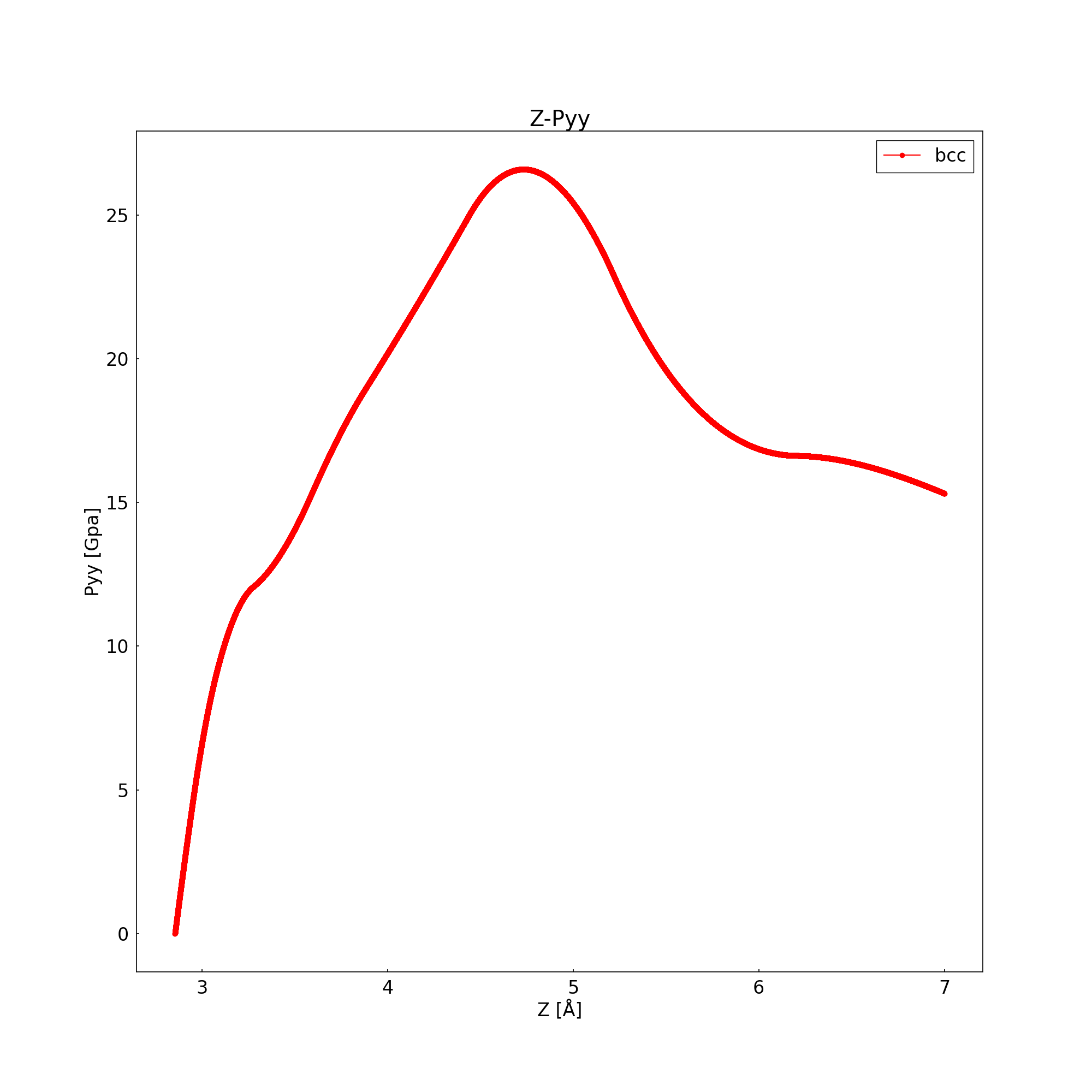
z=2.8553

z=7.0000

variable material string Fe
variable x0 equal sqrt(2)
variable x1 equal 30
variable x2 equal 30
variable x3 equal 30
units metal
boundary p p p
atom_style atomic
lattice custom 1 a1 2.8553 0 0 a2 0 2.8553 0 a3 0 0 hogehogehoge basis 0 0 0 basis 0.5 0.5 0.5
region box block 0 ${x1} 0 ${x2} 0 ${x3}
create_box 1 box
create_atoms 1 box
pair_style eam/fs
pair_coeff * * Fe_2.eam.fs Fe
neighbor 0.3 bin
neigh_modify delay 5
timestep 0.001
# ------------------------ compute pe,ke,stress ------------------------------
compute peratom all pe/atom
compute pe all reduce sum c_peratom
compute kinetic all ke/atom
compute ke all reduce sum c_kinetic
compute stress all stress/atom NULL
compute stress1 all reduce sum c_stress[1]
compute stress2 all reduce sum c_stress[2]
compute stress3 all reduce sum c_stress[3]
compute stress4 all reduce sum c_stress[4]
compute stress5 all reduce sum c_stress[5]
compute stress6 all reduce sum c_stress[6]
thermo 1000
thermo_style custom step lx ly lz press pxx pyy pzz pe c_pe ke c_ke c_stress1 c_stress2 c_stress3 c_stress4 c_stress5 c_stress6
# Setup file output (time in ps, pressure in GPa)
# Output strain and stress info to file
# for units metal, pressure is in [bars] = 100 [kPa] = 1/10000 [GPa]
# p2, p3, p4 are in GPa
variable p2 equal "-pxx/10000"
variable p3 equal "-pyy/10000"
variable p4 equal "-pzz/10000"
variable p5 equal "lx"
variable p6 equal "ly"
variable p7 equal "lz"
variable p8 equal "temp"
variable p9 equal "pe"
variable p10 equal "ke"
variable p11 equal "-pxy/10000"
variable p12 equal "-pxz/10000"
variable p13 equal "-pyz/10000"
variable p14 equal "etotal"
variable p15 equal "vol"
variable steps equal "step"
variable fv equal "sqrt((v_p2-v_p3)^2+(v_p3-v_p4)^2+(v_p4-v_p2)^2+6*(v_p11^2+v_p12^2+v_p13^2)/2)" ##Von Mises Stress
fix equil1 all print 1000 "${steps} ${p2} ${p3} ${p4} ${p5} ${p6} ${p7} ${p8} ${p9} ${p10} ${p11} ${p12} ${p13} ${p14} ${p15} ${fv}" file outfix.txt
# ------------------------ Create custom files with Von Mises Stress for Ovito viewing ------------------------------
variable stress_atom atom sqrt(((c_stress[1]-c_stress[2])^2+(c_stress[2]-c_stress[3])^2+(c_stress[3]-c_stress[1])^2+6*((c_stress[4])^2+(c_stress[5])^2+(c_stress[6])^2))/2)
variable etotal_atom atom c_peratom+c_kinetic
dump out all custom 1 outdmp-pikapikapika.txt id type x y z fx fy fz v_stress_atom v_etotal_atom c_peratom c_kinetic c_stress[2]
run 0
Part 2:
When I noticed this, I change my file. I created a box of Fe(30x30x30). Separate it into group top and bot. And move the group top to draw the fig about move distance and Py.
Q2: Am I right? Because this fig has two peaks and peak value is too small. There is my fig and script:
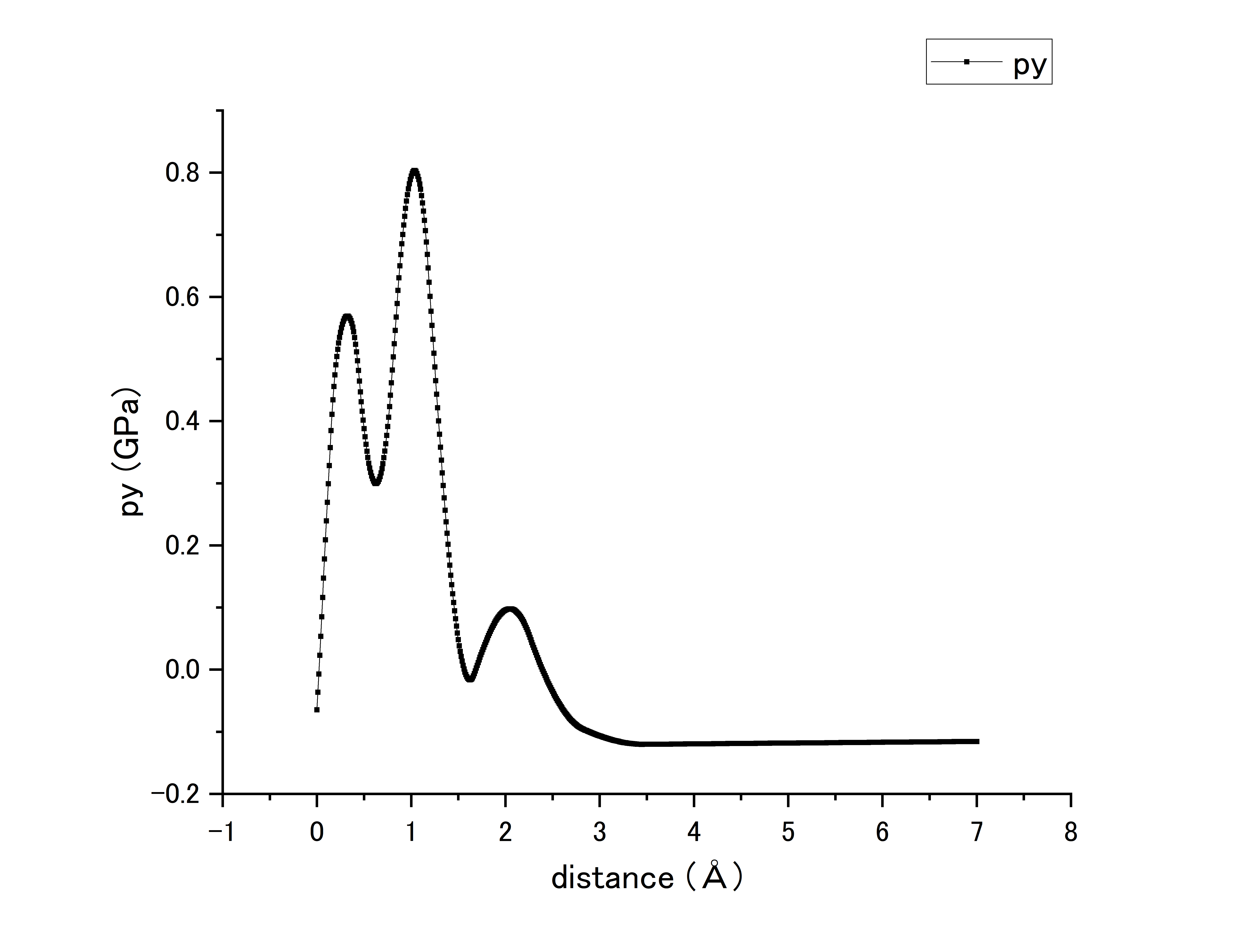
units metal
boundary p s p
atom_style atomic
timestep 0.001
neighbor 0.3 bin
neigh_modify delay 5
lattice bcc 2.8553
region box block 0 85.659 0 85.659 0 85.659 units box
create_box 1 box
create_atoms 1 box
region top block INF INF 42.8295 INF INF INF units box
group top region top
group bot subtract all top
pair_style eam/fs
pair_coeff * * Fe_2.eam.fs Fe
compute 1 top group/group bot
compute peratom all pe/atom
compute pe all reduce sum c_peratom
compute kinetic all ke/atom
compute ke all reduce sum c_kinetic
compute stress all stress/atom NULL
compute stress1 all reduce sum c_stress[1]
compute stress2 all reduce sum c_stress[2]
compute stress3 all reduce sum c_stress[3]
compute stress4 all reduce sum c_stress[4]
compute stress5 all reduce sum c_stress[5]
compute stress6 all reduce sum c_stress[6]
thermo 1000
thermo_style custom lx ly lz press pxx pyy pzz pe c_pe ke c_ke c_stress1 c_stress2 c_stress3 c_stress4 c_stress5 c_stress6 c_1 c_1[1] c_1[2] c_1[3]
# Setup file output (time in ps, pressure in GPa)
# Output strain and stress info to file
# for units metal, pressure is in [bars] = 100 [kPa] = 1/10000 [GPa]
# p2, p3, p4 are in GPa
variable p2 equal "-pxx/10000"
variable p3 equal "-pyy/10000"
variable p4 equal "-pzz/10000"
variable p5 equal "lx"
variable p6 equal "ly"
variable p7 equal "lz"
variable p8 equal "temp"
variable p9 equal "pe"
variable p10 equal "ke"
variable p11 equal "-pxy/10000"
variable p12 equal "-pxz/10000"
variable p13 equal "-pyz/10000"
variable p14 equal "etotal"
variable p15 equal "vol"
variable steps equal "step"
variable fv equal "sqrt((v_p2-v_p3)^2+(v_p3-v_p4)^2+(v_p4-v_p2)^2+6*(v_p11^2+v_p12^2+v_p13^2)/2)" ##Von Mises Stress
fix equil1 all print 100 "${steps} ${p2} ${p3} ${p4} ${p5} ${p6} ${p7} ${p8} ${p9} ${p10} ${p11} ${p12} ${p13} ${p14} ${p15} ${fv}" file outfix.txt
fix 1 top move linear 0.0 0.1 0.0 units box
# ------------------------ Create custom files with Von Mises Stress for Ovito viewing ------------------------------
variable stress_atom atom sqrt(((c_stress[1]-c_stress[2])^2+(c_stress[2]-c_stress[3])^2+(c_stress[3]-c_stress[1])^2+6*((c_stress[4])^2+(c_stress[5])^2+(c_stress[6])^2))/2)
variable etotal_atom atom c_peratom+c_kinetic
dump out all custom 1000 outdmp-*.txt id type x y z fx fy fz v_stress_atom v_etotal_atom c_peratom c_kinetic c_stress[2] c_stress[1] c_stress[3] c_stress[4] c_stress[5] c_stress[6]
run 70000
Part3:
I want to find the ideal strength of the potential energy function I used. If the two ways mentioned above are not correct, I hope to hear your guidance and suggestions.
Many thanks.