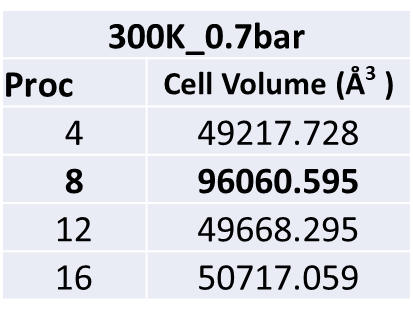
If I change the number of procs (ex. 8 or 16), I take different results. I tried to run flexible materials. I do a calculation with the same input file and the same data file, depending on the number of procs I get different results. Below is a table of the numbers of procs as a function of the final volume. The initial volume of the material is 47000.
This is completely insufficient information. At minimum we need to know what you are simulating and also have a copy of your input script, as well as your LAMMPS version.
This is my input file.
log log.TEST append
neighbor 1.0 bin
units real
atom_style full
boundary p p p
pair_style lj/cut/coul/long 12.5 12.5
bond_style harmonic
angle_style hybrid cosine/periodic fourier
dihedral_style harmonic
improper_style fourier
dielectric 1.0
pair_modify tail yes mix arithmetic
special_bonds lj/coul 0.0 0.0 1.0
box tilt large
read_data data.TEST &
extra/bond/per/atom 10 &
extra/angle/per/atom 10 &
extra/dihedral/per/atom 10 &
extra/improper/per/atom 10 &
extra/special/per/atom 10 &
kspace_style pppm 0.0001
OUTPUT OPTIONS
#settings
thermo 5000
thermo_style custom step temp pe etotal vol density evdwl ecoul emol press
velocity all create 300 2534526 mom yes rot yes dist gaussian
############ HEAT IN NVT #####################
fix 1 all nvt temp 300 1000 100
reset_timestep 0
timestep 1
dump nvtdump all dcd 1000 heat.dcd
restart 50000 heat.*.restart
run 100000
CONSTANT TEMP 1000K
fix 1 all nvt temp 1000 1000 100
restart 50000 heat.*.restart
run 100000
########### COOL THE SYSTEM
fix 1 all nvt temp 1000 300 100
restart 50000 cool..restart
run 100000
###############################################################################################
unfix 1
fix 1 all nvt temp 300 300 100
restart 50000 nvt..restart
run 100000
##############################################################################################
unfix 1
velocity all zero linear
fix 1 all nvt temp 300 300 100
restart 50000 nvt.*.restart
run 100000
undump nvtdump
##############################################################################
unfix 1
timestep 1
reset_timestep 0
fix 1 all npt temp 300. 300. 200 iso 0.70 0.70 2000
fix 2 all momentum 1 linear 1 1 1
dump nptdump all dcd 1000 npt.dcd
restart 200000 npt.*.restart
run 10000000
write_data final.data
END PROCEDURE
If I run with 8 procs and 10000000 steps I took a volume equal to 96060.595. But If I run with 16 procs I took a volume equal to 50717.059.
This is only one of the three things that you were asked to provide and it was done rather badly, since you didn’t quote the file contents correctly. Please see the forum guidelines for instructions.
My Lammps Version is lammps/stable_2Aug2023_update3 and I am simulating MOFs.
Hi @deraptis,
Please have a look at the formatting guidelines of the forum. You will also find there general rule of thumb on how to investigate your issue yourself and communicate the aspect your think are suspicious with regards to LAMMPS use.
It is currently impossible to say anything based on your input since we do not know the system setup or the forcefield. Some settings like box tilt large also seems pointless in isotropic simulation or the fix momentum since there is no external/random forces and the total momentum was already set to 0. When you say you “took a volume” what does it mean? Is it your final value or an average over time? Are you sure your starting system is the same in all case? What is your input command for execution and processor count?
Several issues and bugs were corrected in the last stable releases. Consider updating if possible.
Hi Germain,
When I say “I took a volume”, I mean the volume in the last step. I will upload the data file, there are the force field of my system.
The input command is (mpirun -np #[number of procs] lmp -in in.Test -screen output.out &), if you mean that.
I try to run CH4 adsorption in MOF.
1750 atoms
2064 bonds
3696 angles
5088 dihedrals
3024 impropers
6 atom types
8 bond types
12 angle types
3 dihedral types
1 improper types
0.000000 36.160100 xlo xhi
0.000000 36.160100 ylo yhi
0.000000 36.160100 zlo zhi
0.000000 0.000000 0.000000 xy xz yz
Masses
1 63.546000000 # Cu3+1
2 15.999400000 # O_R
3 14.006700000 # N_R
4 12.010700000 # C_R
5 1.007940000 # H_
6 12.010700000 # C_1
Bond Coeffs
1 57.988603 2.604000 # Cu3+1 Cu3+1
2 190.026252 1.918161 # Cu3+1 O_R
3 646.680887 1.311940 # O_R C_R
4 663.718927 1.345073 # N_R C_R
5 561.491352 1.422196 # N_R C_R
6 391.669513 1.458000 # C_R C_R
7 462.655054 1.379256 # C_R C_R
8 357.440381 1.081418 # C_R H_
Angle Coeffs
1 fourier 68.052595 0.343737 0.374972 0.281246 # Cu3+1 Cu3+1 O_R
2 fourier 152.421295 0.343737 0.374972 0.281246 # O_R Cu3+1 O_R
3 cosine/periodic 74.969428 -1 3 # Cu3+1 O_R C_R
4 cosine/periodic 120.000516 -1 3 # C_R N_R C_R
5 cosine/periodic 110.133094 -1 3 # C_R N_R C_R
6 cosine/periodic 187.136328 -1 3 # O_R C_R O_R
7 cosine/periodic 131.755710 -1 3 # O_R C_R C_R
8 cosine/periodic 102.183280 -1 3 # C_R C_R C_R
9 cosine/periodic 111.297508 -1 3 # C_R C_R C_R
10 cosine/periodic 57.289016 -1 3 # C_R C_R H_
11 cosine/periodic 153.699352 -1 3 # N_R C_R C_R
12 cosine/periodic 141.337441 -1 3 # N_R C_R C_R
Dihedral Coeffs
1 6.737110 -1 2 # Cu3+1 O_R C_R O_R
2 3.368555 -1 2 # C_R N_R C_R C_R
3 1.250000 -1 2 # C_R N_R C_R C_R
Improper Coeffs
1 2.000000 1.000000 -1.000000 0.000000 0 # C_R N_R C_R C_R
Pair Coeffs
1 0.005000 3.113691 # Cu3+1 Cu3+1
2 0.060000 3.118146 # O_R O_R
3 0.069000 3.260689 # N_R N_R
4 0.105000 3.430851 # C_R C_R
5 0.044000 2.571134 # H_ H_
6 0.294100 3.730000 # CH4_sp31
I will take my chances in the lottery here and guess that there is something wrong with your force field or topology. Changing the amount of mpi slightly change the phase space trajectory and thus instantaneous values of properties (this has been discussed here in the forum in at least 2 or 3 different threads that I’ve seen). Probably with 8 processors you reach a "failing’ trajectory, where atoms get too close and start flying away and leading to the expansion of the system. This is maybe why you have such a large volume. Have you tried looking at your last configuration in the case of 8 processors to see how it looks like?
Yes, I have looked at it and the final structure is close to the open pore structure I want to achieve. My force field has worked in similar type of structure.