Dear all
My question here is two fold: one is related to improper style and other is related to vmd topotools generating impropers (dont know whether i should post here or in vmd posts. since questions are related i am posting here).
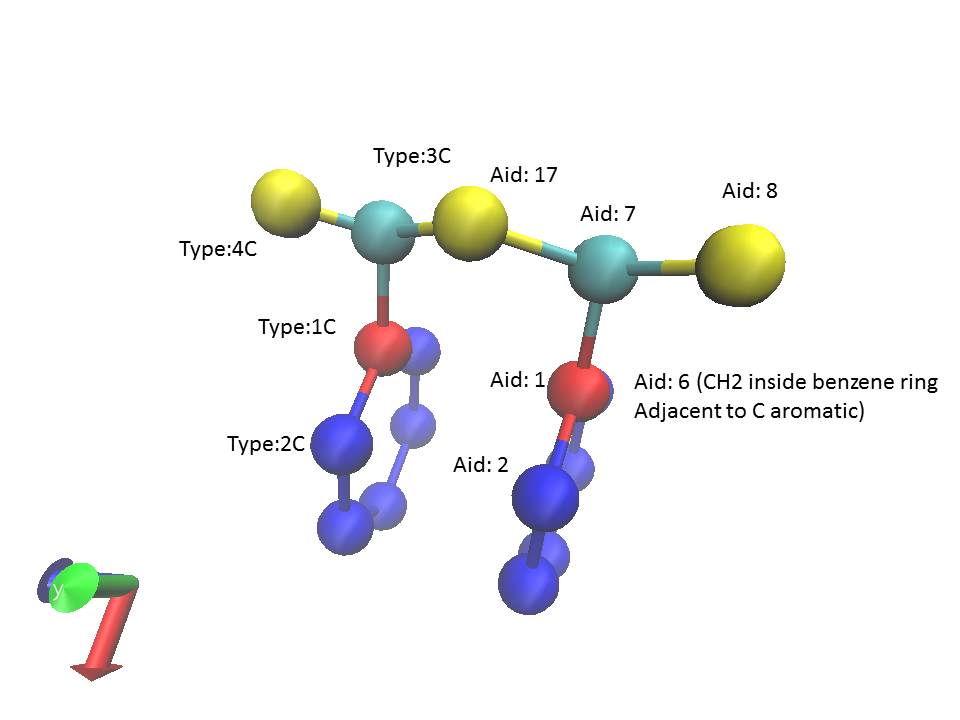
- In LAMMPS manual it is written “Alternatively, you can think of atoms J,K,L as being in a plane, and atom I above the plane, and X as a measure of how far out-of-plane I is with respect to the other 3 atoms.” With reference to the figure which is a simple styrene molecule, (atom ids are given in the figure). I am understanding it as say I want to write improper around atom id 7 (sp3 carbon above the benzene molecule) I would write my improper file as
1 1 7 17 18 1
This would imply that these four atoms form an improper with planes of 17-7-1 and 17-1-8 at some improper angle phi. Is this correct? Or the ordering of the atom ids do not matter?
- After I generate the pdb file and load into vmd. When I do
topo guessimpropers
it is generating an improper between atomids 7-1-6-2. It is written specifically that impropers are calculated when an atom is exactly bonded by 3 other atoms when in a plane. However, if the naming convention is correct, then 7-1, 7-6 and 7-2 are not connected (1-7, 1-2 and 1-6 are however). So again does the order matter? Moreover the type in improper coefficients correspond to reading the improper in reverse order (compare line 52 and 161).
As a follow up question, I want to have impropers to mimic a force field between atom ids 7-17-8-1, i.e, the sp3 carbon above benzene should hold a tetrahedral shape. Is it possible to do in VMD since guessimpropers seems not to find that set?
Thanks alot in advance. Sorry if it is a lot in one single email.
Thanks
Vaidyanathan M S
isocheck.lammps (2.74 KB)