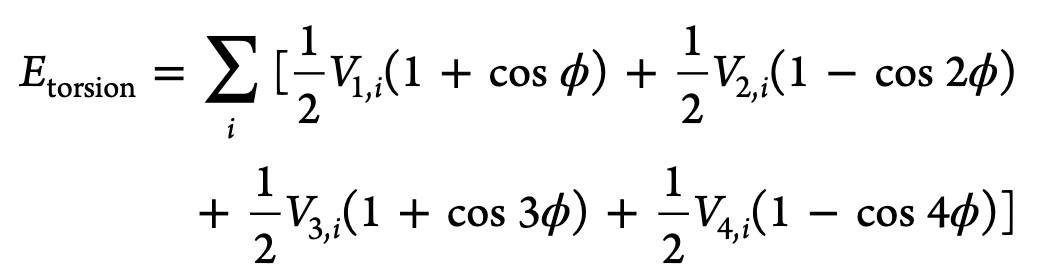Hi!
I’m trying to create a LAMMPS datafile using the OPLS-AA force field for an Ionic Liquid by using the parameters from this paper.
The torsional part of this ff is given as:
X in this context usually means any atom type. The non-X atom type would be the central atom
You could either define them as dihedrals using dihedral style opls or use improper style cvff with d=-1 and n=2 and K=\frac{1}{2}V_2 since the other fourier coefficients are zero.
While writing the impropers in the datafile as “n improper_type I J K L”; would the atom I be CW/CR/NA?
Also, I there are 3 atoms (J,K,L) bonded to I, does the order of writing them?
Hi Alex, could I double check that is the 1/2 term for avoiding double counting, e.g. atom pairs 1234 and atom pair 4321? So in such that the special_bonds lj/coul for OPLS scaling is not applied to improper terms? Many thanks, Catherine
No, just thinking about where the 1/2 term comes from in the (K=1/2 V2). While now got it, it’s from the torsion energy expression and the cvff style doesn’t not include 1/2.
As is discussed in the documentation of most such styles it is entirely a matter of convention whether the this 1/2 factor is explicitly included in the functional form of the potential or implicitly as part of the force constant. Both styles include this factor, just handle it differently, which then means that the force constant must be scaled accordingly if there is a difference between explicit or implicit use of the factor.