My LAMMPS version is 28 Mar 2023. I run the programs on Linux.
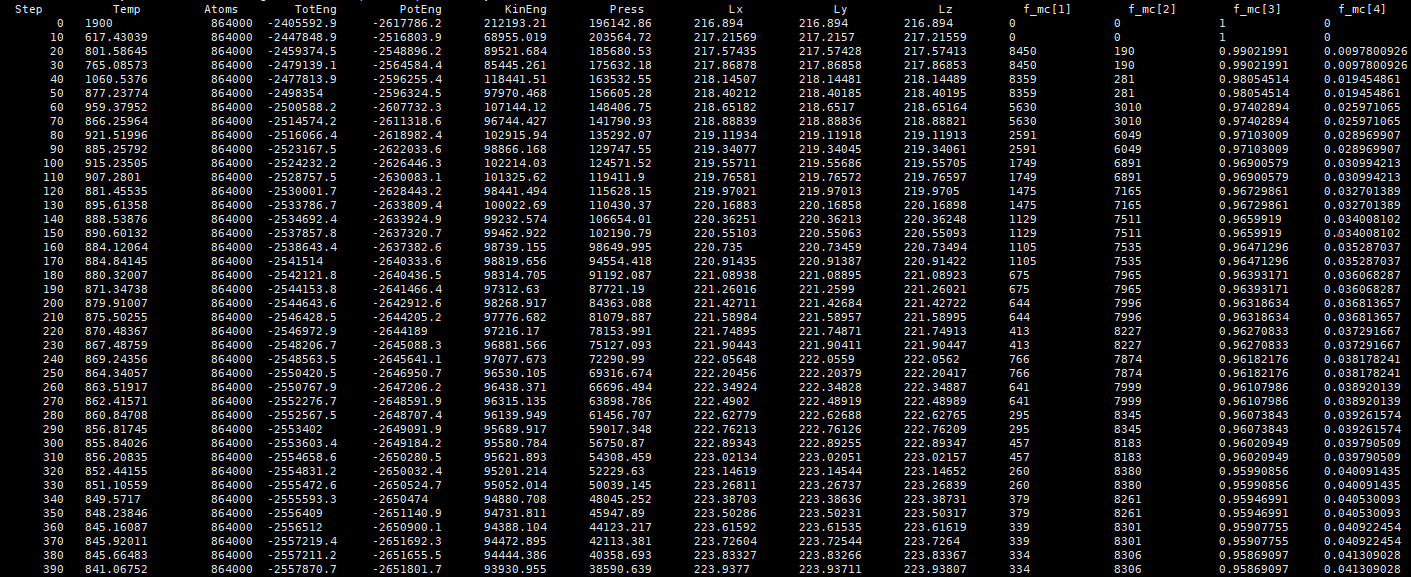
When performing MD/VCSGC simulations, as described in the Scalable parallel Monte Carlo algorithm for atomistic simulations of precipitation in alloys(2012) , when k(kappa) is set to a very small value, the unreasonable chemical potential difference(deltamu) leads to large concentration fluctuations. However, in the simulation of the CuCr system, in order to select a suitable chemical potential difference, I set a value of kappa=1 and changed deltamu for the calculation, and when deltamu<0, the number of exchange successes(f_mc[1]) and failures(f_mc[2]) remained unchanged no matter what the deltamu was,as shown in Figures.
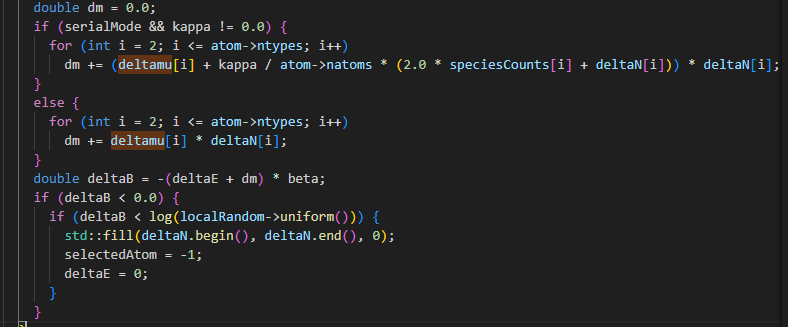
According to the Metropolis acceptance criterion provided by fix_sgcmc.cpp in the source code, deltaB = -(deltaE + dm) * beta, theoretically the smaller deltamu is, the higher the acceptance probability is, and the higher the number of acceptances. In addition, according to the energy difference calculation method provided by Database-driven semigrand canonical Monte Carlo method: application to segregation isotherm on defects in alloys, we calculate ideltaE<0 of CuCr system, So with the setting deltamu<0, the theoretical probability of reception is 1, the number of failures is 0. So, I would like to know why this simulation result occurs.
Here is my running program:
general variables
variable temperature equal 950
variable size equal 60
variable nsteps_run equal 1000000
variables for ‘fix sgcmc’
variable nsteps_mc equal 20
variable swap_fraction equal 0.01
variable temperature_mc equal ${temperature}
variable deltamu equal -2.5
variable target_concentration equal 0.02
variable kappa equal 1
general settings
units metal
atom_style atomic
set up structure
boundary p p p
lattice fcc 3.6149
region box block 0 {size} 0 {size} 0 ${size}
create_box 2 box
create_atoms 1 box
reset_timestep 0
timestep 0.0025
pair_style eam/alloy
pair_coeff * * CuCr.eam.alloy Cu Cr
initialize velocities
variable double_temp equal ${temperature}*2
velocity all create ${double_temp} 428459 dist gaussian
what and how to run
fix integrate all npt &
temp {temperature} {temperature} 1.7 &
aniso 0.0 0.0 1.5
fix mc all sgcmc {nsteps_mc} {swap_fraction} {temperature_mc} {deltamu} &
randseed 324234 &
variance {kappa} {target_concentration}
set up output
thermo 10
thermo_style custom step temp atoms etotal pe ke press&
lx ly lz f_mc[1] f_mc[2] f_mc[3] f_mc[4]
thermo_modify flush yes
dump 1 all custom 10000 mc.*.dump id type x y z
run ${nsteps_run}