Dear all,
I have been trying to compute the Short Range Order (SRO) parameter for a equibinary Mo-Nb system using the neighbor lists provided by LAMMPS. The results outputted are incoherent: the number of NbMo pairs should be strictly equal to the number of MoNb pairs, as we are looping over every atom in the system.
Here is the 2000 atom BCC system, equibinary Mo Nb
NbMo_binary.dat (152.6 KB)
initial_datafile= NbMo_binary.dat
pair_count_dic_list_n1=[]
pair_count_dic_list_n2=[]
NVT_steps_between_SRO= 50 #between each sampling
in_file=f"""
clear
units metal
dimension 3
atom_style atomic
read_data {initial_datafile}
pair_style adp
pair_coeff * * W_Mo_Nb.adp.alloy Nb Mo
neigh_modify every {NVT_steps_between_SRO} check no
fix 1 all nvt temp 700 700 $(dt*100)
"""
lmp.commands_string(in_file)
lmp.command(f'run {NVT_steps_between_SRO} pre no post no')
## build lists of dictionnaries containins the pair counters ##
pair_count_dic_n1=get_pair_count(initial_datafile,lmp,'adp')
pair_count_dic_list_n1.append(pair_count_dic_n1)
pair_count_dic_n2=get_pair_count(initial_datafile,lmp,'adp',n_shell=2)
pair_count_dic_list_n2.append(pair_count_dic_n2)
Using the function get_pair_count:
def get_pair_count(datafile,lmp, pair_style,n_shell=1):
## get atom type in string format to have all the possible pairs##
mass_list=extract_masses(datafile)
all_atom_types= [mass_to_symbol(mass) for mass in mass_list]
all_pair_types=[]
for i,atom_type in enumerate(all_atom_types):
for j,neighbor_type in enumerate(all_atom_types):
#if atom_type!= neighbor_type:
pair = atom_type+neighbor_type
all_pair_types.append(pair)
all_pair_types_dic={}
for i in range(len(all_pair_types)):
all_pair_types_dic[all_pair_types[i]] =0
idx = lmp.find_pair_neighlist(pair_style)
get_atom_type = lmp.extract_atom('type')
# example using NumPy arrays
nplist = lmp.numpy.get_neighlist(idx)
nlocal = lmp.extract_global("nlocal", LAMMPS_INT)
nghost = lmp.extract_global("nghost", LAMMPS_INT)
ntag = lmp.numpy.extract_atom_iarray("id", nlocal+nghost, dim=1)
## get the atom types of the center atom and its neighbors, then count all pair occurences ##
for i in range(0,nplist.size):
atom_i, neighbors_i = nplist[i]
center_atom_id= ntag[atom_i]
center_atom_type=all_atom_types[get_atom_type[center_atom_id]-1]
if nplist.size > 0:
if n_shell==1:
for n in np.nditer(neighbors_i[:8]):
neighbor_global_id= ntag[n]
neighbor_type=all_atom_types[get_atom_type[neighbor_global_id]-1]
print(center_atom_type+neighbor_type)
all_pair_types_dic[center_atom_type+neighbor_type]+= 1
if n_shell==2:
for n in np.nditer(neighbors_i[8:14]):
neighbor_global_id= ntag[n]
neighbor_type=all_atom_types[get_atom_type[neighbor_global_id]-1]
print(center_atom_type+neighbor_type)
all_pair_types_dic[center_atom_type+neighbor_type]+= 1
return all_pair_types_dic
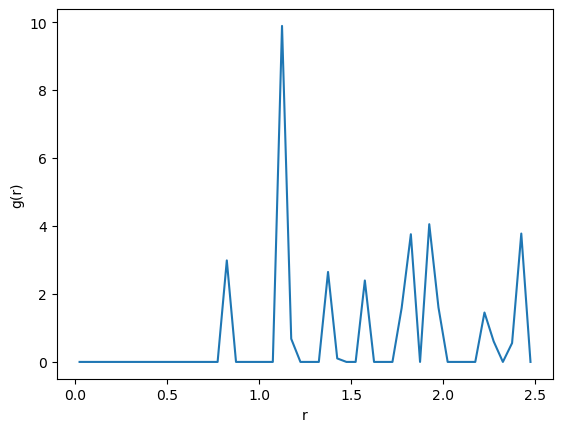
For example one iteration over the system in NVT yields this pair list counter:
{'NbNb': 3929, 'NbMo': 4063, 'MoNb': 4039, 'MoMo': 3969}
This result does not make sense as ‘NbMo’ is not strictly equal to ‘MoNb’.
I could come up with 2 hypotheses that could explain this:
-
Half-list: we might be missing terms due to how the lists are constructed, but I would then assume we would not always get a total number of pairs equal to 16000, which is 8*2000 (the first coordination number for a BCC cell times the number of atoms, which seems to be the case in every attempt.)
-
Behavior of the neighbor lists: I copied the code from a post from akohlmey: Using neighbor list from LAMMPS in Python , but i could not find more documentation on the retrieval of the global ID from the “summation” of the ghost and local IDs.
I hope I have been able to follow the forum guidelines and I thank you in advance for your time and help.
Best,
Olivier