While constructing Pourbaix diagrams (Ti-C system) using Pymatgen, I noticed something strange about the ion energies in the Pymatgen dictionary. The way ion energies are calculated in the code (pymatgen/pymatgen/ext/matproj.py at a1d508844dadbae6d930b66ab88bb13df52bf538 · materialsproject/pymatgen · GitHub) seems logical and consistent for some ions like Ti²⁺ and Ti³⁺. However, when it comes to ions containing carbon (C), the values in the dictionary look unusual.
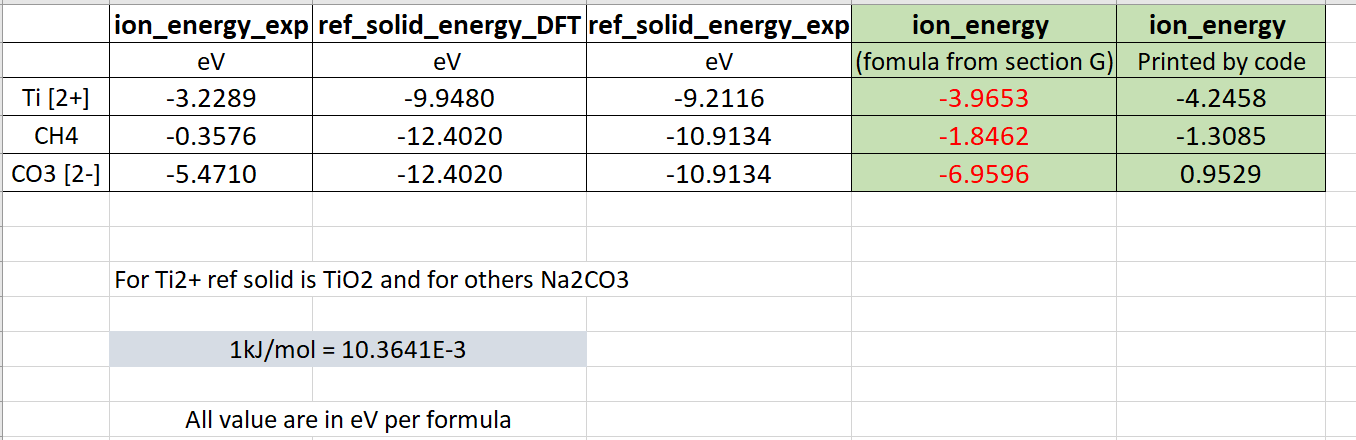
I understand that ion energy is calculated as following.
Ion_energy = (ion_energy_exp ) + [ref_solid_energy_DFT - ref_solid_energy_exp]* (cf) where cf is ratio of major element in ion ion to major element in ref solid.
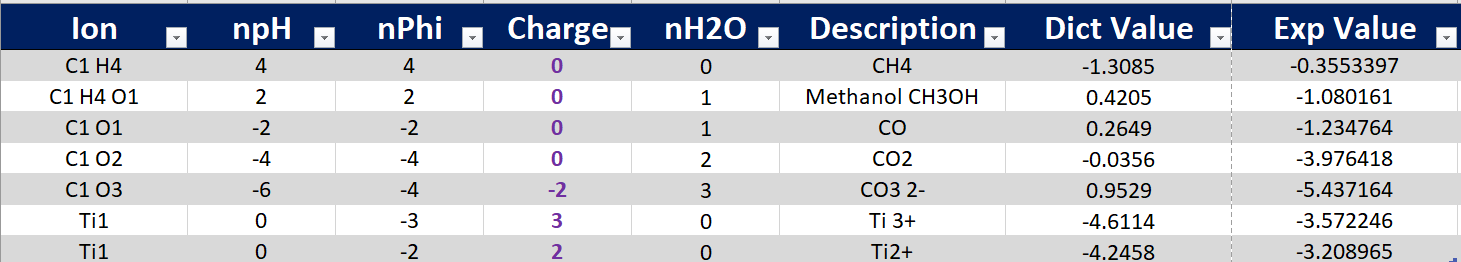
This implies that for the single C-containing entries (such as CH4, CO3-2, CH3OH etc.,) the difference between the dictionary value and the experimental value (available at MPContribs) for each entry is supposed to be a constant. The correction is applied for any C-containing ions using Na2CO3 as the reference solid. So, as long as the ions contain the same amount of C in their formula, the energy difference between the dictionary and experimental values should be the same. But it is not the case.
Could you please highlight the possible reasons for this discrepancy? Thanks.
(Energy in eV/formula)
@rkingsbury