Hello everyone,
I am writing this post because I discovered something strange while performing calculations using VASP.
Let me summarize in two lines:
The data structure received through API in primitive and conventional formats is strange.
When processing these two with BoltzTraP2, a post-processing tool, there are serious value errors. A solution is needed.
Here’s a brief workflow:
- Load structure data from MP_API using Materials Project
- Convert the data using
get_primitive_structure()function to generate a Primitive structure - Separately, using
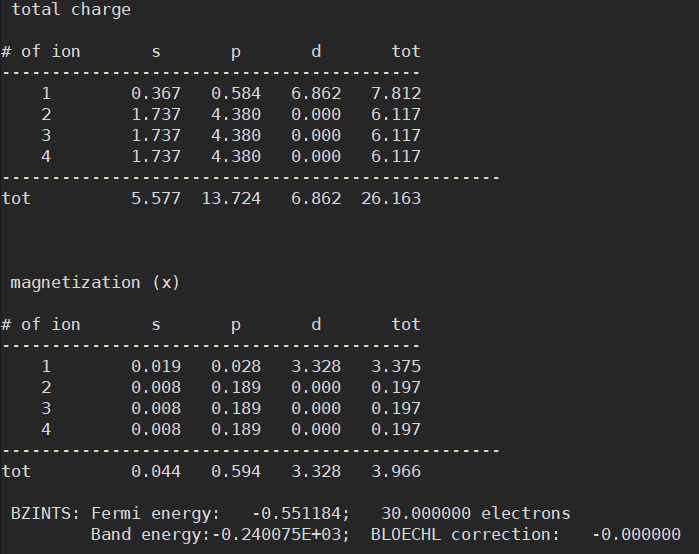
to_conventional()function on the API structure data generates a Conventional structure - At this point, the Final energy (= Total energy; has no physical meaning, but should have values within error range for the same material) from calculations of primitive and conventional structures is confirmed to be the same
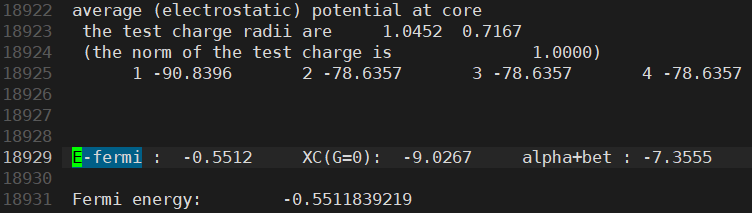
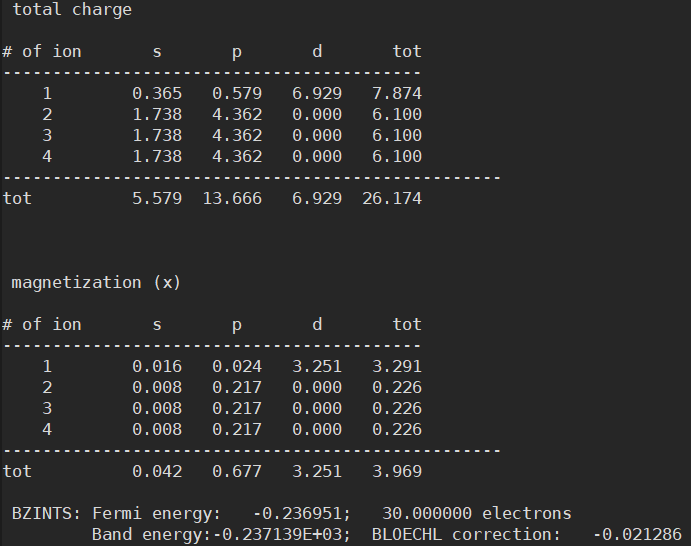
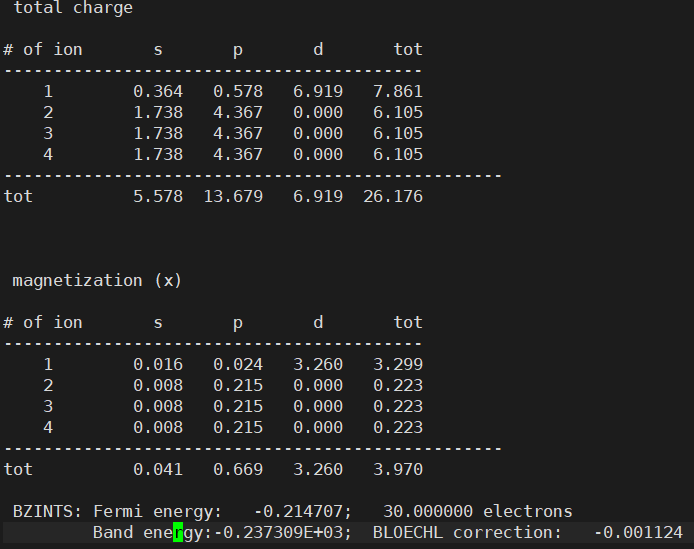
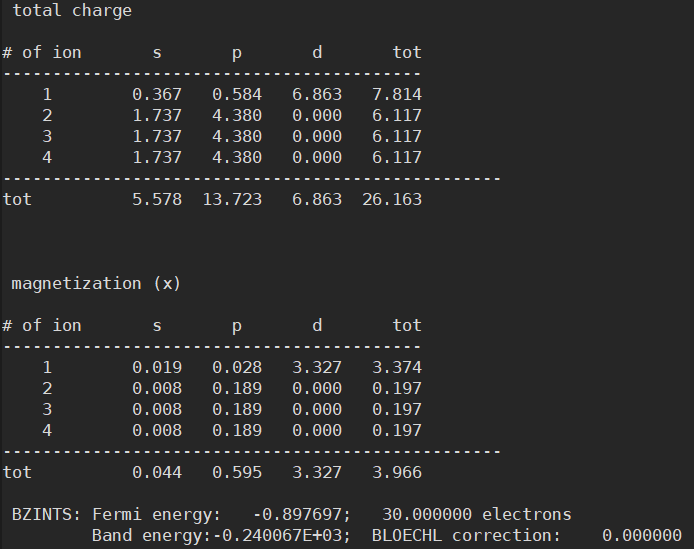
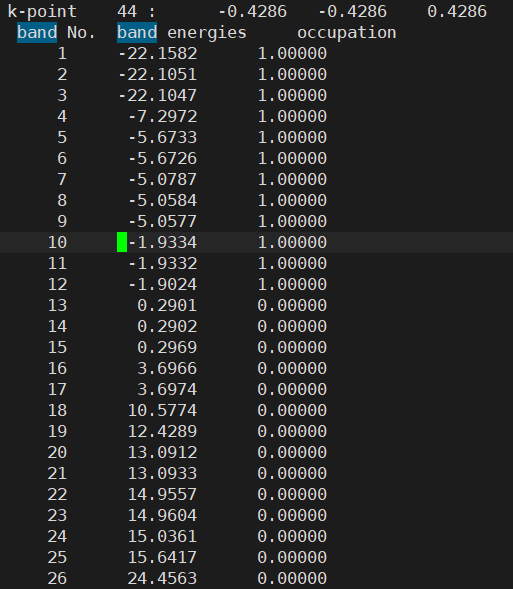
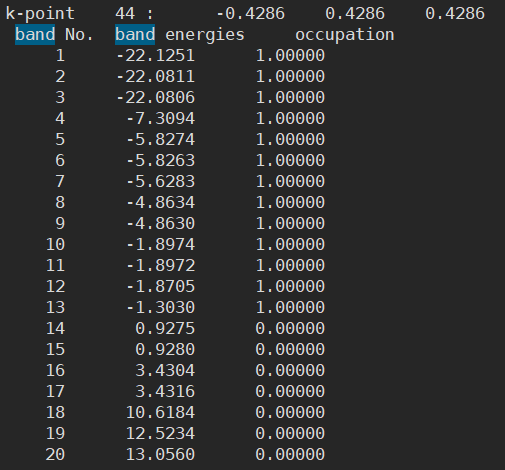
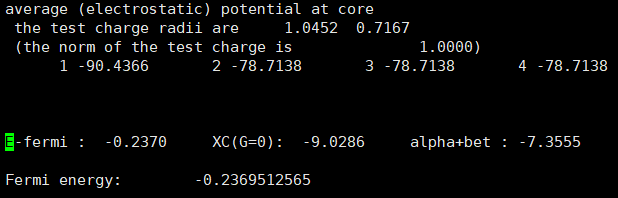
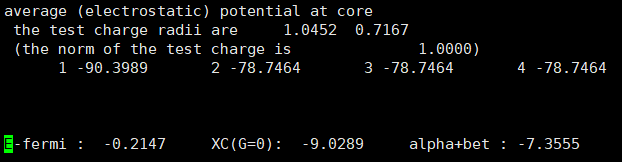
- However, I found that the Fermi energy is different between the two structures
- As a result, the product of resistivity and mean free path obtained during interpolation using BoltzTraP2 comes out very differently. (Since the same material should have the same properties, the values should be the same)
Let me explain in more detail.
First, it seems there is a critical issue with get_primitive.
Previously, when other users and I created topics on similar subjects, the structure obtained through ‘get_primitive’ is quite different from the structure available on the Materials Project website.
The data from the website is actually more similar to the values obtained through ‘to_conventional’.
The structure obtained through ‘get_primitive’ even damages the symmetry of the cell structure.
(e.g., in HCP structure, α, β, γ are not 90, 90, 120, but rather 90, 90, 119.98, etc.)
As a result, calculation errors due to symmetry frequently occurred in both Quantum ESPRESSO and VASP calculations. This was also a common problem in BoltzTraP2, which is used for post-processing.
Anyway, having different Fermi energies in the same structure is indeed problematic. This is because such small differences in Fermi level can cause very large errors in the final results.
get_primitive of Co1F3_mp-559435
- static
- NSCF
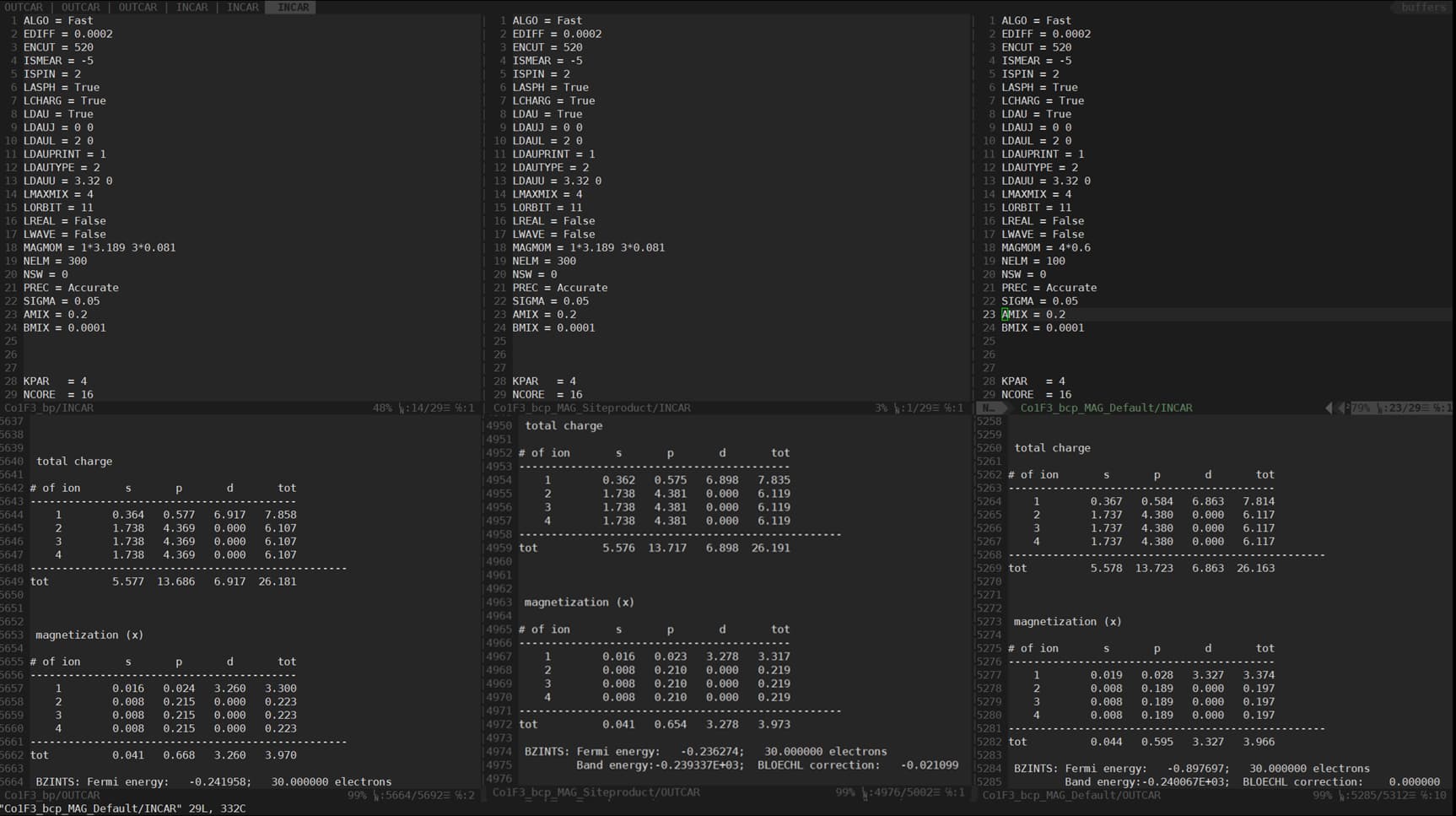
INCAR (Static)
ALGO = Fast
EDIFF = 0.0002
ENCUT = 520
ISMEAR = -5
ISPIN = 2
LASPH = True
LCHARG = True
LDAU = True
LDAUJ = 0 0
LDAUL = 2 0
LDAUPRINT = 1
LDAUTYPE = 2
LDAUU = 3.32 0
LMAXMIX = 4
LORBIT = 11
LREAL = False
LWAVE = False
MAGMOM = 13.189 30.081
NELM = 300
NSW = 0
PREC = Accurate
SIGMA = 0.05
AMIX = 0.2
BMIX = 0.0001
KPAR = 4
NCORE = 12
INCAR(Non-self consistency field)
ALGO = Fast
EDIFF = 0.0002
ENCUT = 520
ICHARG = 11
ISMEAR = -5
ISPIN = 2
KSPACING = 0.05
LASPH = True
LCHARG = False
LDAU = True
LDAUJ = 0 0
LDAUL = 2 0
LDAUPRINT = 1
LDAUTYPE = 2
LDAUU = 3.32 0
LMAXMIX = 4
LORBIT = 11
LREAL = False
LWAVE = False
NELM = 100
NSW = 0
PREC = Accurate
SIGMA = 0.05
KPAR = 4
NCORE = 12
- POSCAR
Co1 F3
1.0
2.6472384033333332 1.5283830066666670 2.1613651899999997
0.0000023733333334 -3.0567679833333337 2.1613661900000003
2.6472397666666665 -1.5283839666666665 -2.1613641800000001
Co F
1 3
direct
0.9999965233333332 0.9999965233333331 0.0000034766666666 Co3+
0.5000006766666666 0.0001207133333332 0.0001185800000000 F-
0.0001207133333334 0.9998814199999999 0.4999993233333332 F-
0.9998810866666666 0.5000006766666668 0.9998789533333333 F-
to_conventional of Co1F3_mp-559435
- INCAR: static
ALGO = Fast
EDIFF = 0.0002
ENCUT = 520
ISMEAR = -5
ISPIN = 2
LASPH = True
LCHARG = True
LDAU = True
LDAUJ = 0 0
LDAUL = 2 0
LDAUPRINT = 1
LDAUTYPE = 2
LDAUU = 3.32 0
LMAXMIX = 4
LORBIT = 11
LREAL = False
LWAVE = False
MAGMOM = 4*0.6
NELM = 300
NSW = 0
PREC = Accurate
SIGMA = 0.05
AMIX = 0.2
BMIX = 0.0001
KPAR = 4
NCORE = 4
- INCAR: NSCF
ALGO = Fast
EDIFF = 0.0002
ENCUT = 520
ICHARG = 11
ISMEAR = -5
ISPIN = 2
KSPACING = 0.05
LASPH = True
LCHARG = False
LDAU = True
LDAUJ = 0 0
LDAUL = 2 0
LDAUPRINT = 1
LDAUTYPE = 2
LDAUU = 3.32 0
LMAXMIX = 4
LORBIT = 11
LREAL = False
LWAVE = False
NELM = 100
NSW = 0
PREC = Accurate
SIGMA = 0.05
KPAR = 4
NCORE = 4
- POSCAR
Co1 F3
1.0
-2.6472393272282879 -1.5283843381846158 2.1613651866667190
-2.6472393272282879 1.5283843381846154 -2.1613651866667194
0.0000000000000000 -3.0567686763692312 -2.1613651866667190
Co F
1 3
direct
0.9999999999999999 0.0000000000000001 0.0000000000000001 Co3+
0.0001198116666665 0.5000000000000001 0.0001198116666667 F-
0.9998801883333335 0.9998801883333335 0.4999999999999999 F-
0.5000000000000000 0.0001198116666666 0.9998801883333335 F-