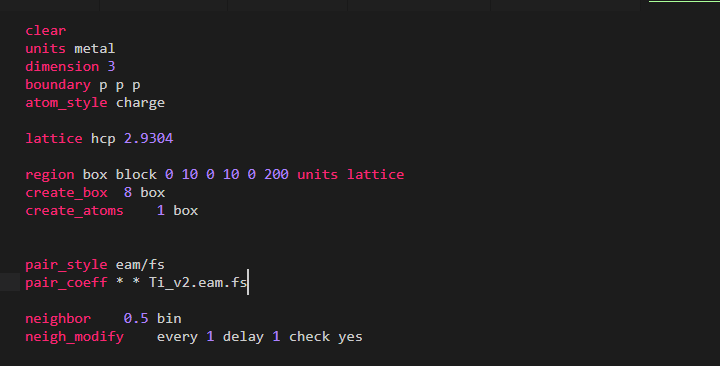
I need to model titanium for a lammps simulation. But the used potential file is causing error.Following is written in the input script,
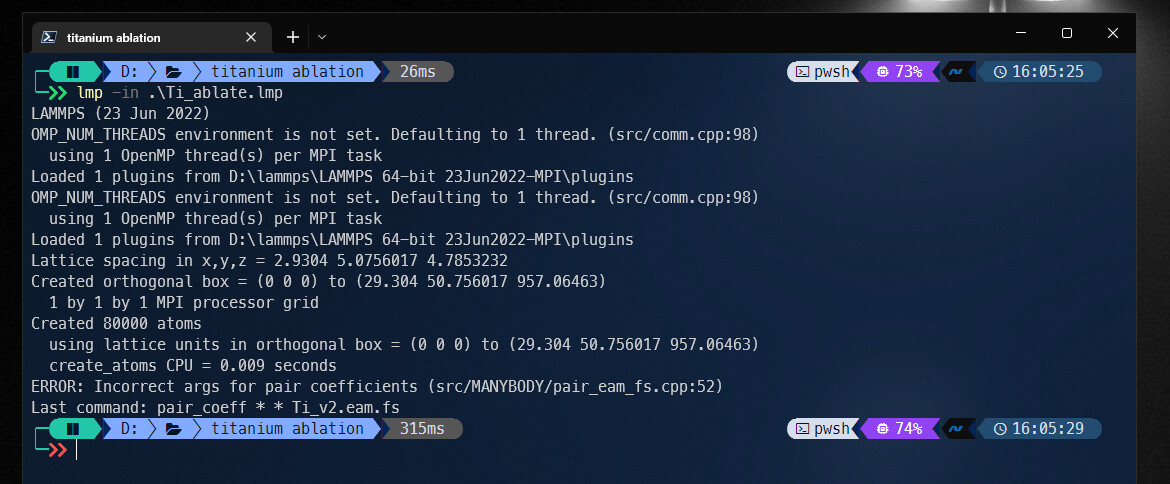
and this is the error i get,
I opened the cpp file indicated in the error (line 52)
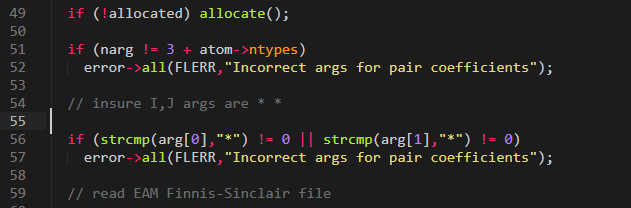
the first two args are * and then the potential file, same as in the input script
this is the file structure
The paper from which I am modeling, adopted a manybody potential

I downloaded the following file
https://www.ctcms.nist.gov/potentials/Download/1992--Ackland-G-J--Ti/3/Ti_v2.eam.fs
This is written in lammps format.
If someone could point out the source of error I would really appreciate it.
TIA