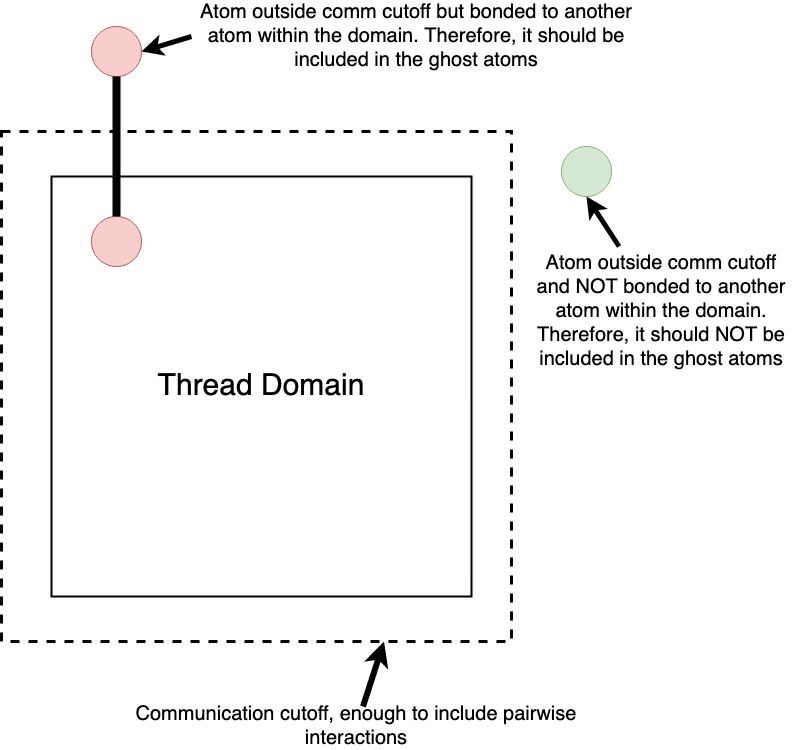
I am dealing with a particular case where the bonds between atoms are generally much longer than the pairwise interaction cutoff. As a result, I have to significantly increase the communication cutoff in order to take into account these long range bonded interactions. Of course, this increases computational costs, as much more ghost atoms are included per thread. However, most of these ghost atoms are totally useless, since they can only interact with the domain’s “true” atoms only through short-range pairwise interactions.
Given the above, I am basically trying to find a way to draw just the ghost atoms that interact through bonds with the domain atoms. I have included an image that may help a bit in explaining what I am trying to do:
Do you think that there may be a way to implement something like that in LAMMPS? As fas as I have understood from the documentation, there is no way to selectively create ghost atoms after a certain range (maybe it is possible through creating groups? Still, I cannot use this approach in my case because it would require creating more groups than the upper limit allowed by LAMMPS)
That was my first thought as well. The problem is that there is only one atom type in my case, i.e. all atoms have the same pairwise and bond interactions. On the image of my original post one should imagine that the green particle is also bonded to other particles, its just that those other particles do not belong to the domain in question. To put it another way, I want to set a different communication cutoff regarding the bonded interactions, not the atoms per se.
Excuse me if my question sounds ignorant, I made sure to check the documentation but I am still a bit fuzzy on the details of ghost atom communication between threads.
Then you are out of luck. If all atoms are bonded and bonded with the same bond type, then you must include all in the communication cutoff. If you have the same interaction parameters but different bonded/non-bonded status, you can just use multiple atom types with the same parameters.
Beyond that, it may be possible but will require some effort programming a new multi communication setting. But before that, we first need to see an example that can be used to quantify how much of a benefit there will be. It may not be worth the effort.
It is even more restrictive. You not only want to restrict to bonded atoms, but to bonded atoms where the bond straddle the subdomain boundary.
There is no ghost atom communication between threads, only between MPI ranks.
Exactly. I understand that this is probably asking too much from a piece of software that is designed to be user-friendly in order to facilitate non tech-savvy users like myself.
It probably isn’t worth the effort but I am attaching a minimal example in case you are interested in having a look. To give you some context, I am basically trying to compress a system of aggregates, where each aggregate is “rigidified” by bonds between its constitutive particles. Particles belonging to the same aggregate may be quite far apart, thus increasing the maximum bond length of the system. Other than that, the particles interact only through excluded volume.
Not necessarily. A lot of the code in LAMMPS was written by people that needed to solve a particular problem. Apart from a few fundamental design decisions, the code base allows for rather diverse usage patterns, and there are many applications that people have implemented which were rather unexpected by the (core) LAMMPS developers.
In this case, for example, the straightforward approach to implement this, is to utilize the comm_modify multi code path, by adding a different method to define “collections” (there are basically two categories/collections of atoms: bonded to a local atom with one communication cutoff based on the maximum bond length and the others with a default communication cutoff). That already has two strategies implemented: one suitable for discrete element modeling where the particle radius varies a lot, and one suitable for force fields where the pair styles have different cutoffs (e.g. for modeling of colloids, or systems using long-range electrostatics where few atoms have charges and thus is would be more efficient to increase the real space cutoff to reduce the cost of the reciprocal space calculation in PPPM, which primarily scales by volume and not by number of particles).
However, to go about implementing that, one would need a suitable use case. Ideally, you would implement this yourself. Otherwise, you could add a feature request issue on github: Issues · lammps/lammps · GitHub, but that would need a lot of luck of somebody looking at this and deciding that that would be a project with sufficient “hack value”. But, again, a good motivation and a suitable, simple test case, would be key.
If you don’t care about too much about performance and scaling to a large number of processors, you can look into pair style list, which was devised to facilitate a somewhat similar problem: a coarse grained model of DNA or RNA which required very long bonds between strands and where the communication cutoff would prohibit the use with bond styles, even when being able to utilize different atom types and comm_modify multi.
When realizing long bonds with pair style list, all bonds must be listed within the “list” that the pair style processes and not in the data file. The upshot is that it does not require any ghost atoms, since the contribution from the individual bonds are computed where the contributing atoms are local at the expense of doing global communication, which will limit parallel efficiency across many processors.
Thank you for taking the time to provide such a detailed answer. The concept of pair_style list sounds interesting for my case and I will check it out soon. In any case, this discussion has been very enlightening on some aspects of LAMMPS that I am not so familiar with.