Hello everyone,
I tried to simulate a tensile test on CsI3Sn, but when I ran my script, the temperature reduced from 300K to 0 immediately and I couldn’t take my result.
Please help me to solve my problem.
00ss.in (1.9 KB)
CsI3Sn_33750.data (1.2 MB)
You need to learn to be better at debugging your simulation input yourself.
A few pointers:
- Never ever use
thermo_modify lost ignore unless you have a simulation where atoms are supposed to leave the system. This is obviously not the case here
- Do not discard certain thermo output, unless you know you can safely do it
- If a simulation has problems, try using a shorter time step, more frequent thermo output, and create a trajectory dump with correspondingly frequent output
- Remember the GI-GO principle (= garbage in, garbage out). This means, if any of your simulation settings are badly chose, chances are high that your simulation will crash or produce bad results. Thus look at all parameters and think about whether they make sense
- Always double check whether unit conversions were done correctly
Applying these techniques will show you that your system does not go to “0 K” instantly. It also shows that you are losing atoms and that you have a peculiar behavior with your potential energy, which makes sense when looking at your potential parameters which in turn confirms that this is a GI-GO case.
Thank you for your help. It was very helpful for me.
But when I remove this command, I get this error:
ERROR: Lost atoms: original 33750 current 33748 (…/thermo.cpp:481)
Exactly, so there is a big problem with your simulation in that atoms get accelerated so much that they get lost. This is a very useful hint indicating that your overall input has big problems.
Problems do not get solved by ignoring or hiding them, but by understanding the cause and addressing them.
Remember me mentioning the GI-GO principle. So now you need to figure out whether each and every bit of your input is correct and meaningful. I would start with the pair style settings and parameters. They look extremely questionable to me.
I fixed it by setting a shorter time step. (0.0001 ps)
But the temperature dropped to zero again.
I’m trying your ways to solve this issue.
and the reference for pair coefficients is:
https://doi.org/10.1016/j.enganabound.2022.11.032
That does not fix anything, only confirms my suspicion.
That is behind a paywall and thus not accessible. Regardless, figuring out how to correctly enter potential parameters is your job and not something that is a LAMMPS problem.
I converted potential parameters as follows:
pair_style lj/cut 2.5
pair_coeff 1 1 0.0242 4.39 1.39
pair_coeff 2 2 0.0143 4.50 1.38
pair_coeff 3 3 0.0017 4.02 2.57
and it is the table of that reference:
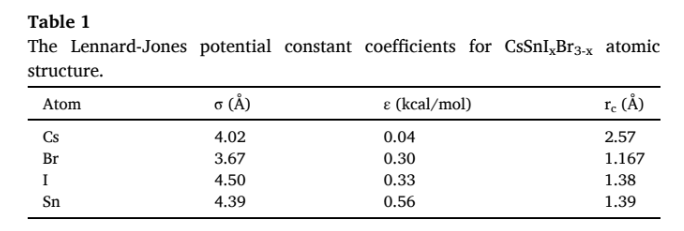
And again the temperature decreased to 0 K instantly.
When I setting a shortet time step, the temperature increases.
Is this a standard, unmodified Lennard-Jones potential? Does r_c represent the cutoff as is implied by your pair_coeff statements?
It makes no sense to have a cutoff much shorter than the Lennard-Jones \sigma parameter as that would result in a potential that is either zero or highly repulsive, which is causing the behavior that you see.
Unfortunately, I don’t know about this.
Excuse me, Is it true to set the LJ cutoff equal to: 2.5 * sigma ?
If you don’t understand this, you must not do simulations until you know enough about it.
Please go and find someone that can teach you and get some text books. You just wasted a whole lot of your and my time.
I mean there is no more information in that article for this simulation.
Thank you anyway. Your guide was very useful for me.