Hello,
Resently i used the Interface Reactions app to calculates some reactions,and i found that cannot be agreed with the result i used pymatgen api.
So i test some reactions and find that the Interface Reactions and Reaction Calculator App results are inconsistent ,too.
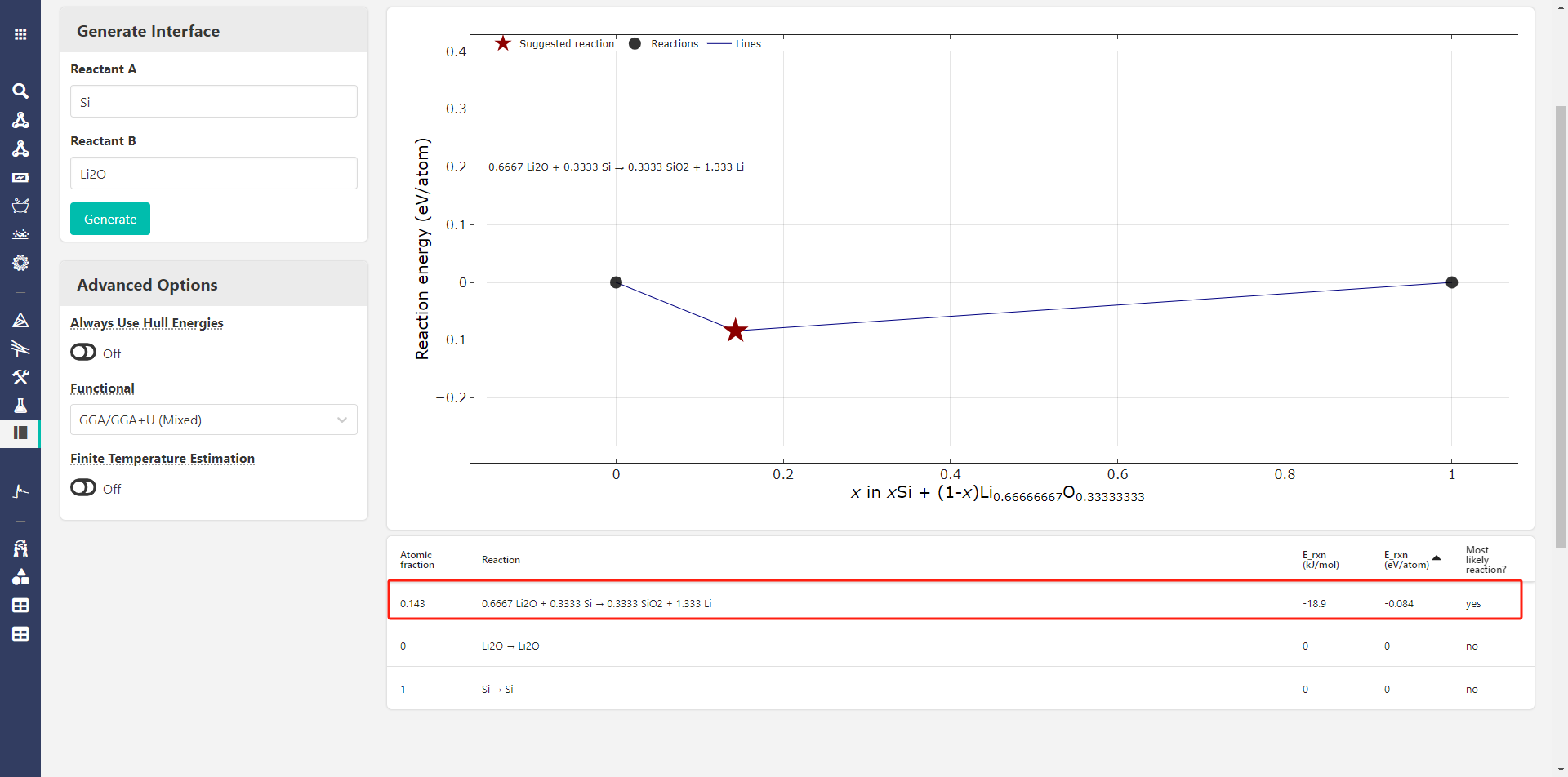
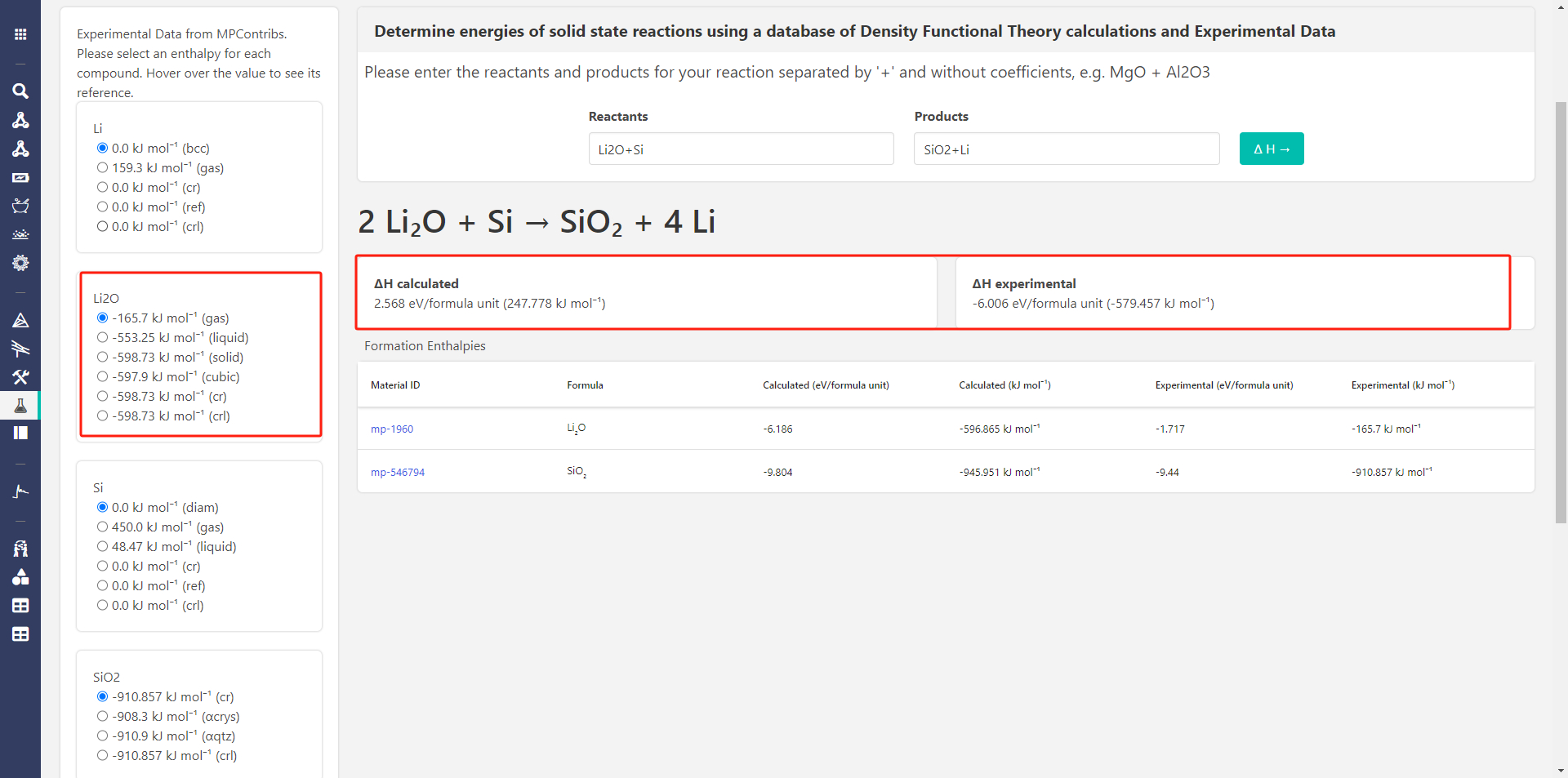
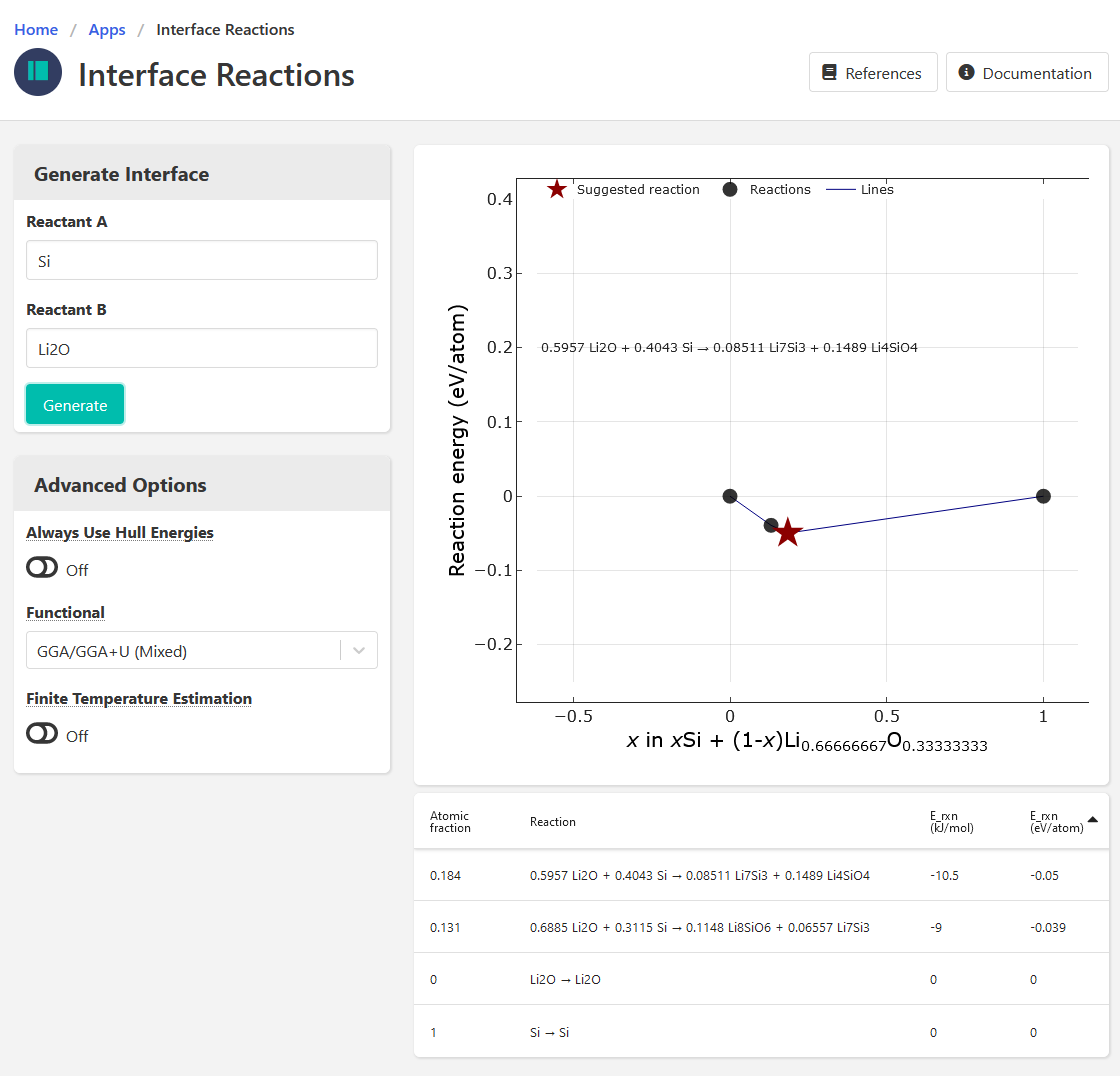
When using Si and Li2O as reactants, the Interface Reactions app give me only one posible reaction:
0.6667 Li2O + 0.3333 Si → 0.3333 SiO2 + 1.333 Li
and it has a negative E_rxn
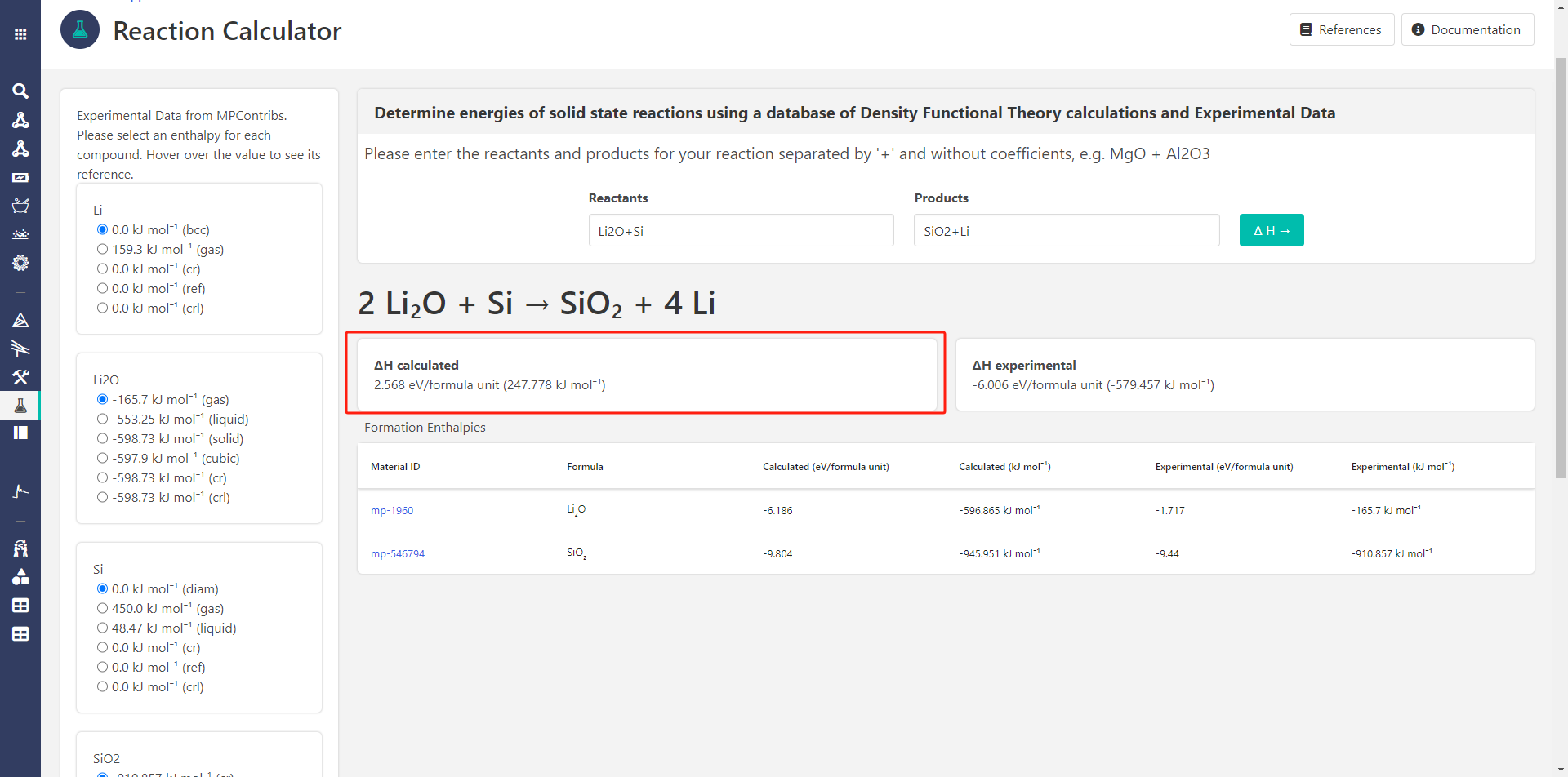
But when i use Reaction Calculator to calculate the same reaction, the calculated ∆H is always postive.
and i find that the experimental ∆H can sometimes become negative.
I wander if the Interface Reactions used the experimental information to calculate ?
And if it true,can i get these information by mp-api ?
Thanks
The Reaction Calculator uses the experimental information shown here or here. Use the mpcontribs-client python library to download the data (see docs). E.g.
from mpcontribs.client import Client
client = Client(apikey="your-api-key-here", project="experimental_thermo")
data = client.download_contributions()
Thanks for your reply.
But this still cannot solve my problem.
I know that Reaction Calculator uses the experimental information and now I will try to use the information to calculate the Interface Reactions.
My problem still here:
When using Si and Li2O as reactants, the Interface Reactions app give me only one posible reaction:
0.6667 Li2O + 0.3333 Si → 0.3333 SiO2 + 1.333 Li
and it has a negative E_rxn
when i use Reaction Calculator to calculate the same reaction, the calculated ∆H is always postive.
Tagging @rkingsbury here who might be able to help
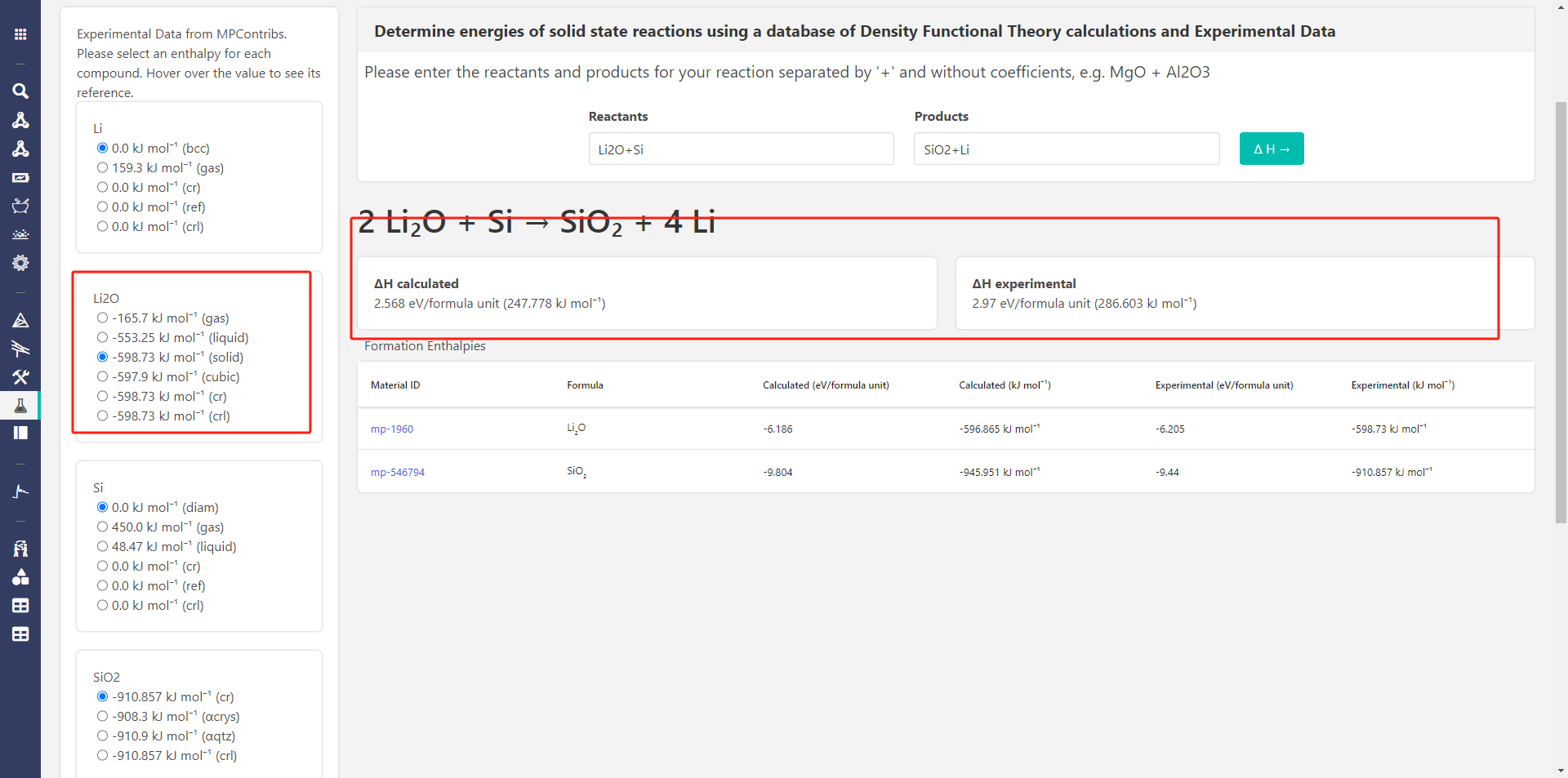
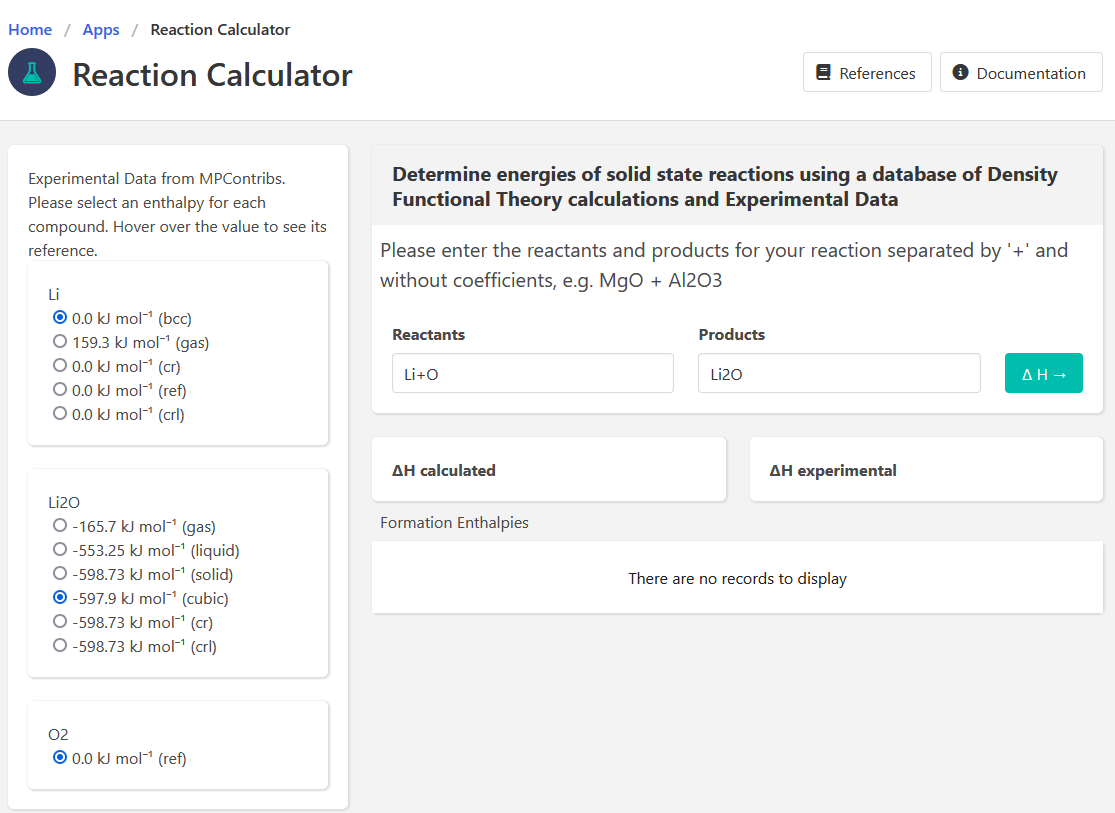
Hi @zzyfor2019 , one thing I notice is that in the screenshot you posted from the Reaction Calculator app, you have selected a the Li2O gas phase as the experimental energy (in the left column). I think you probably want the solid phase, right? If you select that, are the answers more consistent?
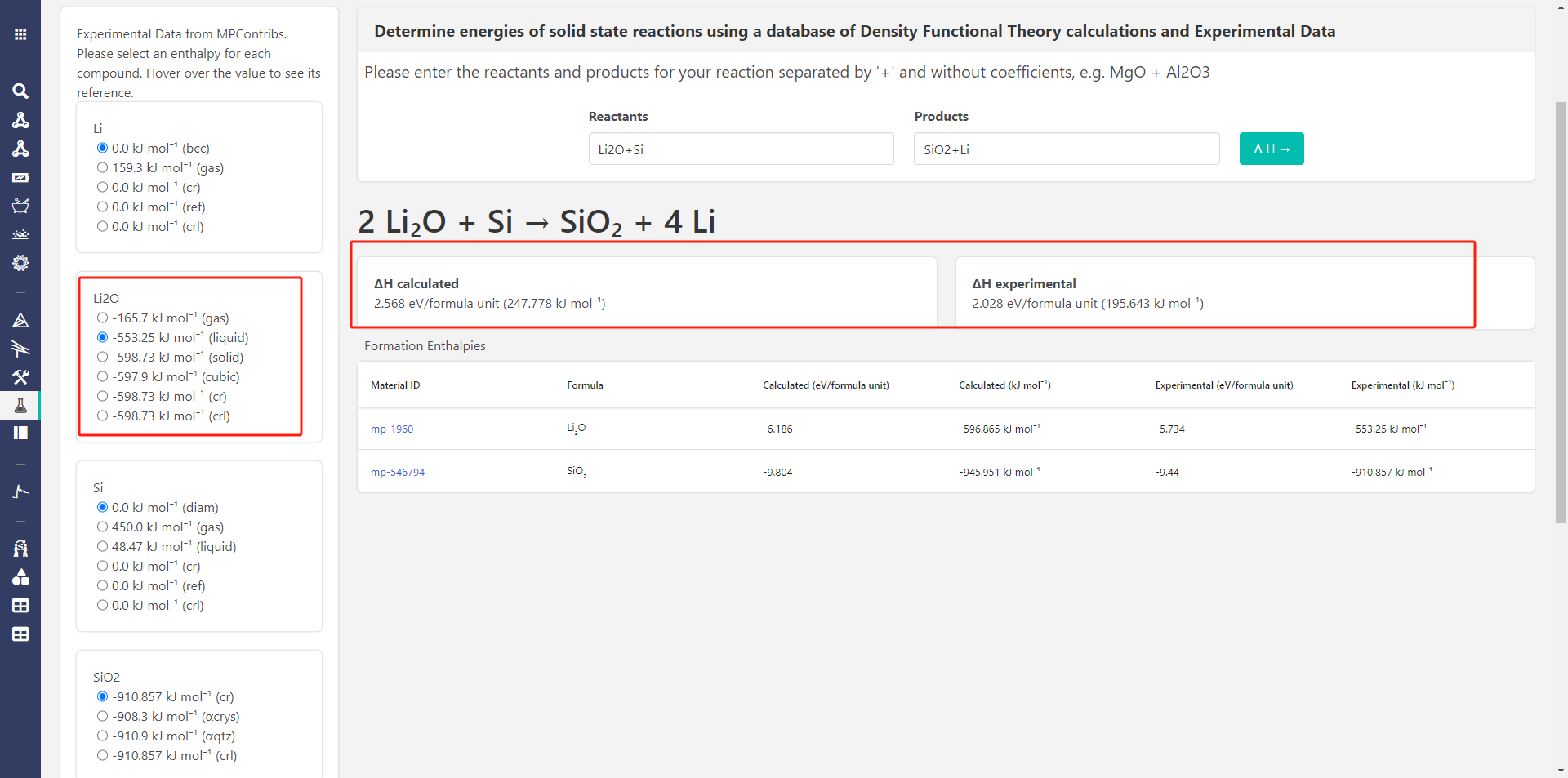
Hi,@rkingsbury, I have selected all the options but they shown the same calculated result , it seems that the options just influence the experimental result.And put them here.
Hi @zzyfor2019 , thanks for checking that.
I am familiar with the Reaction Calculator but not the Interface app. Do you know if both of these use the same experimental data? (@tschaume - do both pull from the MPContribs collection?) Are they supposed to calculate the same energy? (e.g., is the interface app supposed to show a surface formation energy rather than a bulk one?)
Next things to check would be
- Verify that the experimental data in MPContribs is correct (I remember Jordan B. discovered that some of the values had gotten permuted a year or two ago, I think he fixed it though)
- which app has the more accurate energy (I suspect its the interface app)
- Whether there is an error in the way one of the two apps calculates reaction energy
Thanks @tschaume ; sorry for my slow reply here. I saw something in meeting notes recently that there was a problem between legacy and next-gen interface app that was fixed recently; is that related?
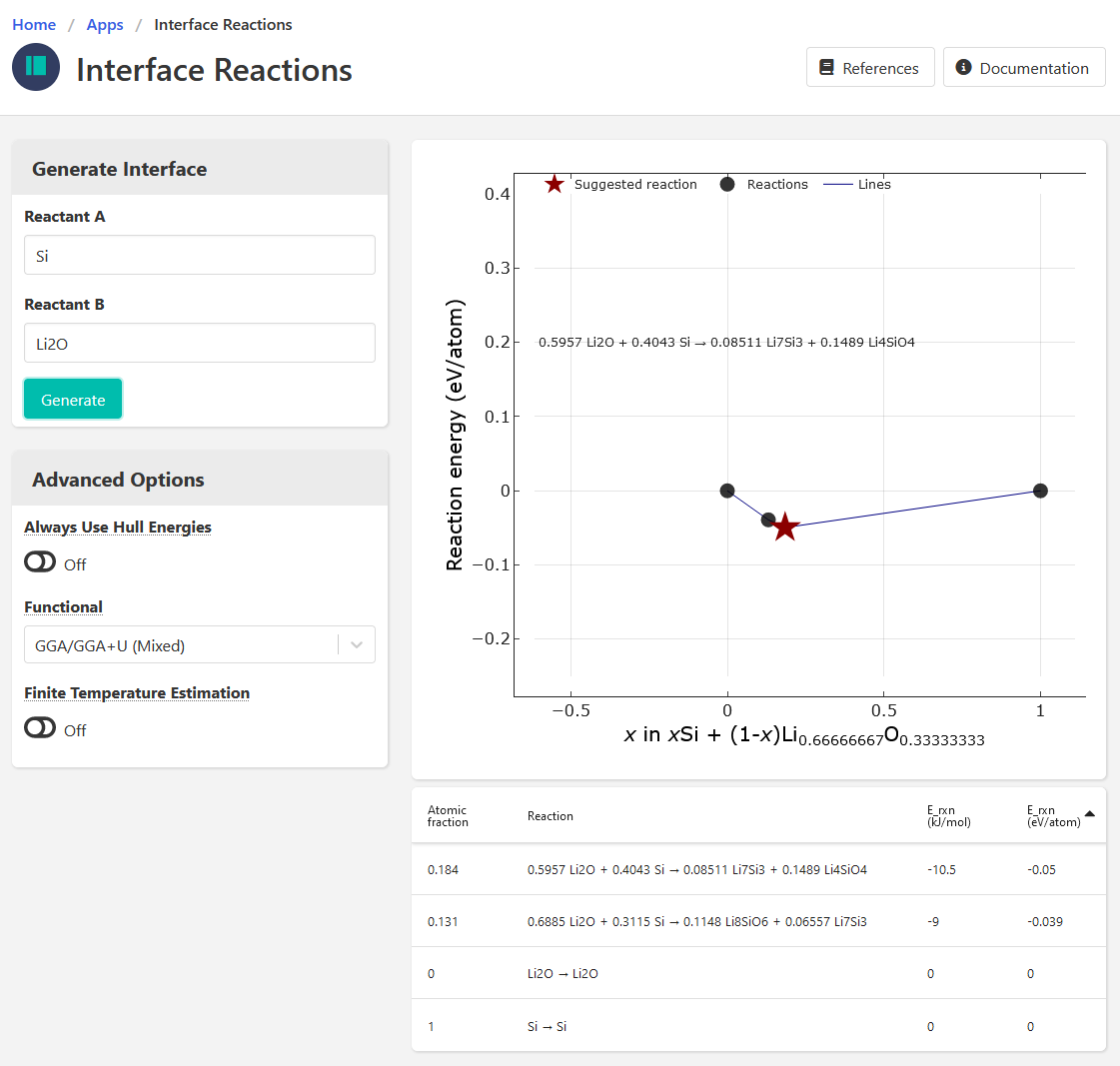
So this morning I went and compared the reaction calculator, interface, and phase diagram outputs for this chemical system - screenshots below. I noted the following:
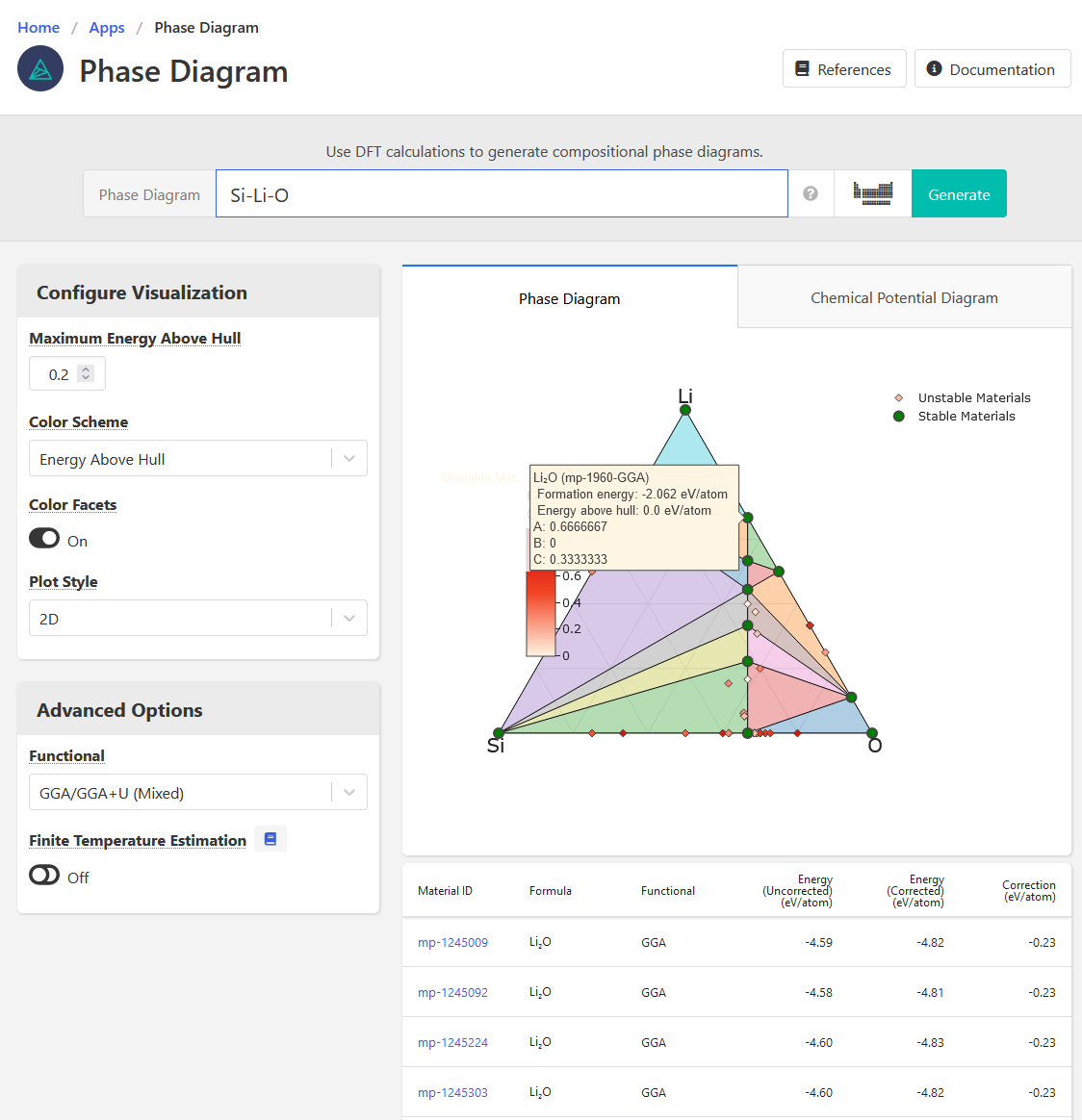
- Formation energy of Li2O is negative on the phase diagram app
- Interface app results in different reactions, but that might be expected since I think those are based on surface energies (But again, I’m not too familiar with this app).
- On the phase diagram, there is no tie line between Li2O and Si, which makes me think the interface app must be using different (surface) energies rather than bulk DFT energies
- In the reaction calculator, I am not able to generate any results for the formation reaction of Li2O. I wanted to check and see if the app would return the same formation energy as the Phase Diagram (which it should). See last image.
@rkingsbury Thanks for looking into this! Yes, @ruoxiyang was working on the interface app recently and her updates went live on MP last week. @ruoxiyang Do you think these issues should be resolved now?
The fix that went out a few weeks ago was related to the first question @zzyfor2019 posted, i.e. there were not enough reactions sourced. That was fixed.
However, the reaction calculator (calculated enthalpy) and the interface calculator are based on different analysis modules: Reaction and Interface calculator, respectively.
In particular, the former uses the calculate_energy() method to get the reaction enthalpy, whereas the latter uses the get_kinks() method to find the lowest formation energy pathways. And it becomes slightly more complicated because experimental values are involved in the Reaction Calculator. These two apps are
We should improve the documentation of these two apps to avoid confusion.