The new interface reactions apps looks awesome! Congrats on putting that out today.
Out of curiosity, will there be a way to implement this via PyMatGen? Currently, pymatgen.analysis.interface_reactions.InterfacialReactivity() looks like it’s close to the MaterialProject website app, with the exception of the phasediagram input. In the app, the phase diagram is generated automatically, whereas pymatgen’s InterfacialReactivity() requires a self-generated phase diagram as an input. It’d be very useful if pd=None defaulted to the algorithm used in the website app.
Or is there a quick function in pymatgen.analysis.phase_diagram that already generates the desired pd input for us…?
Here is an example Python workflow to compute interface reactions using MP data via pymatgen. The notebook is a bit out of date – some data fields changed names and so later code cells may error without minor changes – but the first four cells (building the phase diagram and obtaining the reaction data) should still work. We will update this notebook soon to reflect the current version.
Specific to your question (and detailed in the linked-to notebook), the method MPRester.get_entries_in_chemsys is key to gather the data for pymatgen’s PhaseDiagram constructor.
Oh, I realize that you are probably also interested in pymatgen’s MPRester.mpr.get_interface_reactions method, which automatically does what you want using MP data.
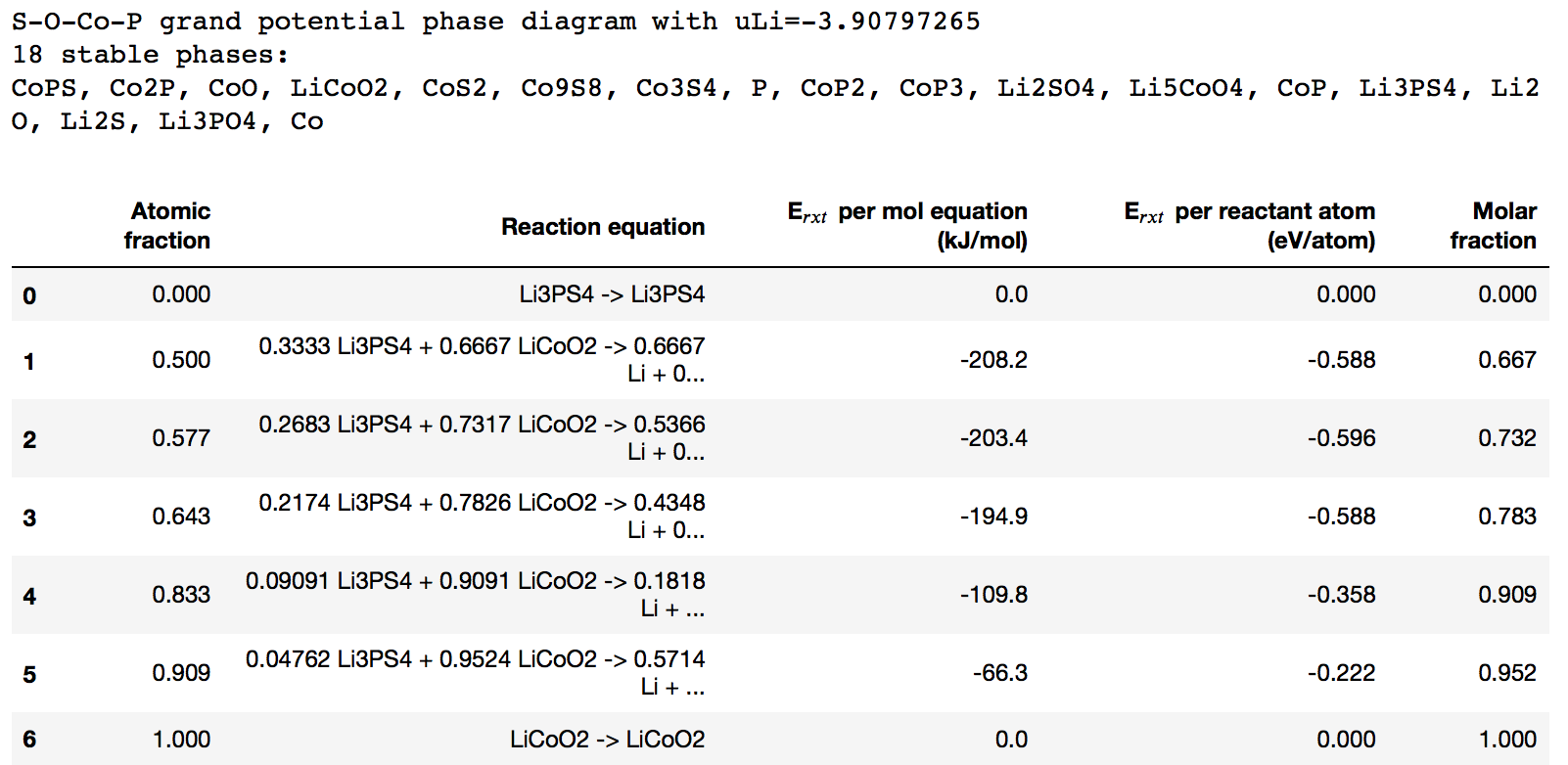
May I bother you or anyone here with the Interface Reaction App in Open system? I am really confused in determining the chemical potential of the open element. I did similar for LiCoO2 and Li3PS4 reactants using the MP Interface Reaction code (version 2019.2.28). I chose Li as the open-element with input mu_Li=-2. However, when I check the mu_Li in output, its value is mu_Li=-3.9.
I think the value of mu_Li is important in calculating reaction energy (Er) for open system. For example:
0.2683 Li3PS4 + 0.7317 LiCoO2 -> 0.5366 Li + 0.2439 Co3S4 + 0.09756 Li2SO4 + 0.2683 Li3PO4
Er(eV/atom)= {0.5366mu_Li + 0.2439Ef[Co3S4] + 0.09756Ef[Li2SO4] + 0.2683Ef[Li3PO4] - 0.2683Ef[Li3PS4] - 0.7317Ef[LiCoO2]}/[0.2683x8 + 0.7317x4]
Even I use mu_Li=-2 or mu_Li=-3.9, I can’t get the same Er=-0.596 (eV/atom) as shown by the Interface Reaction app. Is there anything wrong with my reaction energy calculation or determining the mu_Li?