Dear Everyone,
I have a question on calculating force on an atom in a group that I specified. My simulation is for polymers.
Please see the figure attached for the detail question description.
Is there a command available in Lammps or a simple way for calculating the resultant internal force on an atom ONLY from the other atoms in the same group?
Thank you for any commons,
Lili Zhang
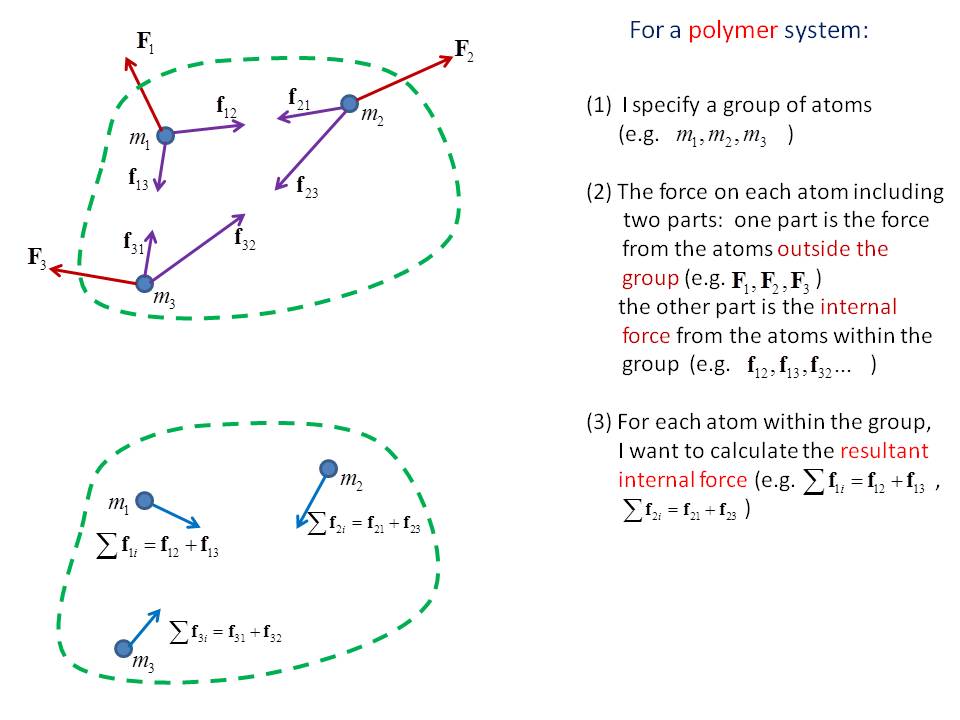
Dear Everyone,
I have a question on calculating force on an atom in a group that I
specified. My simulation is for polymers.
Please see the figure attached for the detail question description.
Is there a command available in Lammps or a simple way for calculating the
resultant internal force on an atom ONLY from the other atoms in the same
group?
rather than asking about a technical detail,
can you explain what you want to use the
breakdown of forces like this for?
you cannot have simple access to individual
force components, since there are too many
and they are just summed up. it might be possible
to do something in postprocessing, however,
before going on tangents, it may be easier to
discuss the overall goal first and perhaps find
an out what it is that you really need to do.
cheers,
axel.
Dear Dr. Kohlmeyer,
Sorry for not explaining my goal clearly.
My ultimate goal is to define our own stress tensor for a specific group of atoms.
To do this, for each atom in the group, I need to calculate the resultant force which is only from all the other atoms in the same group onto the atom under consideration.
After such internal force calculation, I will use them for our stress calculation for this group of atoms.
Although I know there is command available in Lammps for stress calculation, I can’t use them directly. Since our stress is based on a different perspective, this is the reason for such internal force calculation.
Thank you very much,
Lili Zhang
University of Nebraska-Lincoln
Dear Dr. Kohlmeyer,
Sorry for not explaining my goal clearly.
My ultimate goal is to define our own stress tensor for a specific group
of atoms.
To do this, for each atom in the group, I need to calculate the resultant
force which is only from all the other atoms in the same group onto the
atom under consideration.
After such internal force calculation, I will use them for our stress
calculation for this group of atoms.
Although I know there is command available in Lammps for stress
calculation, I can't use them directly. Since our stress is based on a
different perspective, this is the reason for such internal force
calculation.
i don't see how you could do this without
modifying the c++ code in lammps.
to do it the way you describe it, would
require quite a few changes, in fact,
depending on how you would identify
which atoms would belong to the
same group.
there may be a smarter way to do this
by directly accumulating individual
stress tensor components similar to
how it is done already and just skipping
over the parts you don't want.
but that is difficult to tell with the
available information. i guess you'll
have to dig into the code and figure it out.
perhaps steve has an idea?
axel.
I think you can use compute group/group with one
of the groups as a set of atoms and the other group
as one atom in the set.
Steve
Thanks Steve.
Is there a limit on the total number of groups that I can define in the compute group/group ?
I didn’t find this information in the doc page.
Lili
The limit on the number of groups within LAMMPS is 32.
That has nothing to do with the compute group/group command.
If what you really want is to define a group and have the force
on each atom in the group due only to other atoms in the group,
then compute group/group is not a good solution. As Axel said,
LAMMPS doesn't tally any such statistics. You could write a compute
to do it, I suppose, if it needs to be done on the fly. Or you
could post-process your dump file with a rerun command,
and use the neigh_modify exclude options to effectively mask
out all the atoms not in the group, and recompute the forces
you want for each snapshot.
Steve
Or you
could post-process your dump file with a rerun command,
=)
This is a nice example why the "rerun" command is so useful.
and use the neigh_modify exclude options to effectively mask
out all the atoms not in the group, and recompute the forces
you want for each snapshot.
Incidentally, if for some reason you have trouble turning off the
forces with neigh_modify, you can take a more radical approach. You
could create a new data file and/or input script which turns off all
forces between atoms which are not in the target group (including
3-body, and 4-body forces if necessary) and use that when use use
"rerun". This allows you to compute energies and stresses using
totally different force-field settings than what you used when you ran
the simulation.
(When you re-analyze your data with "rerun", be careful not to
accidentally use a restart file to initialize the state of your
virtual simulation. Old restart file will contain many of the old
force-field settings. Use the new data file with the new settings
instead.)
rerun is pretty cool.
Andrew
Thanks Steve and Andrew.
I like the "rerun " idea. But my big concern is that the "neigh_modify exclude " command only turns off the non-bond pair interactions, which means I will still have the bond, angle, torsion… interactions between the two groups.
So that for one atom in group1, I am not able to get the internal force only from atoms in group1 onto the atom, since there is still bond interactions from group2 on the atom.
Lili
Thanks Steve and Andrew.
I like the "rerun " idea. But my big concern is that the "neigh_modify
exclude " command only turns off the non-bond pair interactions, which means
I will still have the bond, angle, torsion... interactions between the two
groups.
So that for one atom in group1, I am not able to get the internal force only
from atoms in group1 onto the atom, since there is still bond interactions
from group2 on the atom.
Lili
You can turn them off too. This is what I was getting at in my previous email.
You can always create a new set of input files which turn off any
interactions you want to ignore (including bonds, angles, dihedrals,
many-body, etc). When you use rerun, load the new data/input file(s)
and those interactions will not be included in any computes you
calculate. The coordinates of the atoms will be read from the dump
file generated during your previous simulation, but the computed
stresses or other quantities will be different.
Cheers
Andrew
Dear Andrew ,
Thank you very much.
I will check it out. It will save me a lot of time if this method works.
Have a nice weekend,
Lili Zhang
University of Nebraska-Lincoln, Lincoln, NE 68588-0526