Hello!
I am plan to use AdsorbateSiteFinder on two matched surface slabs to construct the potential energy surface (PES) of the interface.
I would like to first find only the unique sites of the two slabs, and actually compute the energy at all combinations of these points (e.g. on-top_1 / hollow_1, on-top_2 / hollow_1, …)
I already figured out a way to then match the unique points with all the points (returned when symm_reduce = 0). I then have the full unit cell filled with data and can interpolate to get a smooth PES.
In testing this I played around with graphene on Ni111 because of the good match and found some unexpected things:
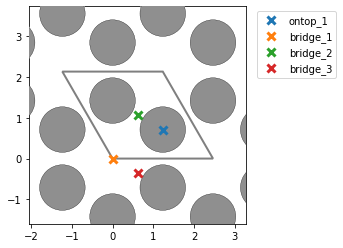
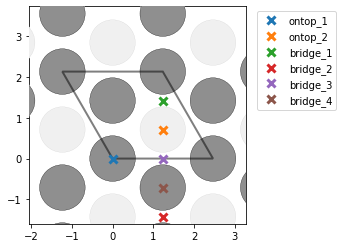
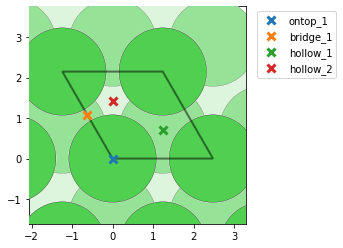
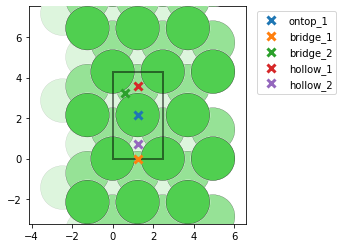
- I find too many unique points for graphene (mp-1040425), while I get the correct amount for graphite (mp-48). Could this be due to there being only one layer in graphene? I see that the unit cells are different, and in this post @Joseph_Montoya explains that sometimes too many sites are listed, but I am a little confused nevertheless why the results are different for the two cells. Here are some pictures to illustrate with graphene on the top (note that bridge_5 is apparently obscured behind another bridge site) and graphite on the bottom:
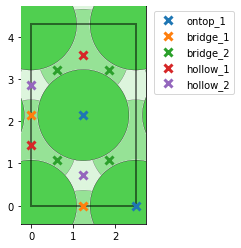
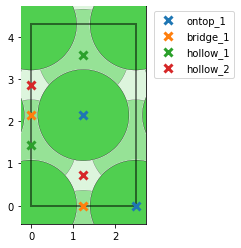
- I am also surprised that the number of unique points changes based on the unit cell of Ni111. If I construct a slab out of the equilibrium fcc structure, i correctly find 1 on-top, 2 hollows, and 1 bridge site. However, if I match the cells and get a new rectangular cell, I get an additional bridge site which seems identical. Could this be due to the small strain that is imposed on the cell during the matching with the graphene cell?:
- If indeed the two bridge sites found in the matched cell are not equivalent, is there an option of telling
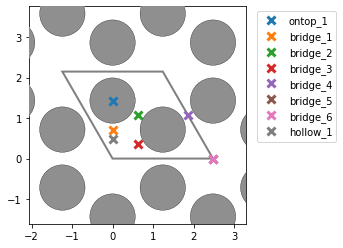
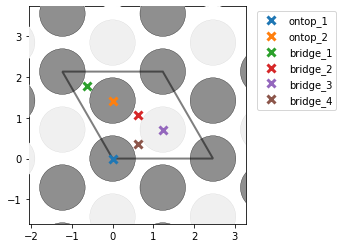
AdsorbateSiteFinderto consider them equivalent? I tested slowly varying bothsymm_reducefrom 0 to 1 andnear_reducefrom 0 to 1 while keeping the other value at the default of 0.01. I found that for both parameters, a value between 0.26 and 0.33 results in the correct number of sites for the unique points, but when I try to look at all points (settingsymm_reduce=0andnear_reduce=0.30; right picture below) I am missing 4 of the bridge-sites in the middle of the cell (compared to settingsymm_reduce=0andnear_reduce=0.01; left picture below).
- My main questions are probably:
- What do
symm_reduceandnear_reduceactually do? Why are the results the same if I change the two parameters independently? The documentation is unfortunately not very helpful there… - Is there a dependence on the chosen unit cell or am I experiencing some effect of small strains that are recognized by the algorithm?
- What do
Thanks for any help, if needed I can also provide some more context or code that I am using!
Cheers, Michael
 I cannot find the old code that I used to produce the pictures and now I cannot reproduce the results myself. I do not think that this has been resolved by an update of pymatgen either, since I am using version v2020.10.20 right now, and in the changelog, nothing for AdsorbateSiteFinder shows up since v2019.7.30. I think that somewhere in the post-processing code (to shift the points back into the unit cell, assign names, etc.) there must have been a bigger bug that I mistakenly attributed to a problem with AdsorbatSiteFinder. We have changed quite a bit since October, and found other challenges, but this problem seems to have been not a problem at all. Thanks for bringing this to my attention!
I cannot find the old code that I used to produce the pictures and now I cannot reproduce the results myself. I do not think that this has been resolved by an update of pymatgen either, since I am using version v2020.10.20 right now, and in the changelog, nothing for AdsorbateSiteFinder shows up since v2019.7.30. I think that somewhere in the post-processing code (to shift the points back into the unit cell, assign names, etc.) there must have been a bigger bug that I mistakenly attributed to a problem with AdsorbatSiteFinder. We have changed quite a bit since October, and found other challenges, but this problem seems to have been not a problem at all. Thanks for bringing this to my attention!