What is the difference between building a crystal using the lattice command and building it just by using read_data?
Hello,
The lattice command just defines a lattice, which can be used latter to place the atoms (with create_atoms). The read_data command reads an existing data file which usually already contain the position of the atoms.
So to answer the title of your question: you can build a crystal within a LAMMPS script using the lattice + create_atoms command, or you can create a data file using an external tool (e.g. moltemplate, python) and then read it using read_data.
Best,
Simon
Institute for Computational Physics
University of Stuttgart
https://simongravelle.github.io
They are two different paths to define your system. Once the system is defined, however, it does not matter how the atoms and box and settings got there.
1 Like
Thanks so much for your informative answer.
I set up a lattice using read-data for gamma glycine. I ran a quick minimization and up on that, I did an equilibrium run with both NPT and NVT ensembles. However, it seems that there are smooth changes in the lattice parameters. then the simulation terminated due to “Out of range atoms - cannot compute PPPM”
Here is my input file:
equilibration run
neighbor 2.0 bin
neigh_modify every 1 delay 10 check yes
thermo_style custom step temp density etotal press cella cellb cellc evdwl
thermo 100
timestep 2.0
velocity all create 298 123987 dist uniform
dump trj_full all atom 1000 trj_full.lammpstrj
dump trj_nvt all atom 1000 trj_nvt.lammpstrj
fix fxnvt all nvt temp 298.0 298.0 2000 # iso 0.986923 0.986923 1000
run 1000000
unfix fxnvt
undump trj_nvt
dump trj_npt all atom 1000 trj_npt.lammpstrj
fix fxnpt all npt temp 298.0 298.0 2000.0 iso 0.986923 0.986923 1000.0
run 2000000
unfix fxnpt
fix fxnpt all npt temp 298.0 298.0 2000.0 aniso 0.986923 0.986923 1000.0
run 2000000
First lines of the output:
0 298 1.4201596 -66385.739 4407.8484 48.508295 53.760165 38.530534 -4565.3863
100 256.81698 1.4201596 -66054.691 -7941.3424 48.508295 53.760165 38.530534 -5089.4892
200 246.49038 1.4201596 -65980.213 20.415518 48.508295 53.760165 38.530534 -5851.8121
300 263.19785 1.4201596 -65955.99 4472.8727 48.508295 53.760165 38.530534 -5983.7592
400 251.78777 1.4201596 -65860.921 376.42231 48.508295 53.760165 38.530534 -6070.2577
And the last lines of the output:
1891100 295.75449 1.5741314 -64709.861 -1128.9125 46.872135 51.946862 37.230919 -7122.5486
1891200 297.09659 1.5722473 -64703.392 387.2654 46.890851 51.967605 37.245785 -7097.0663
1891300 296.69784 1.575907 -64687.062 1275.8099 46.854525 51.927345 37.216931 -7031.7372
1891400 297.07705 1.5737756 -64665.242 -334.51564 46.875668 51.950777 37.233725 -7234.8855
1891500 297.39323 1.5699878 -64647.978 -439.43954 46.913335 51.992523 37.263644 -7172.8097
1891600 294.19531 1.5717885 -64620.115 -1671.324 46.895413 51.972661 37.249409 -7091.3372
1891700 297.3152 1.5731751 -64607.171 -101.73895 46.881631 51.957386 37.238461 -7136.3883
1891800 296.56385 1.5701796 -64606.703 720.97952 46.911424 51.990405 37.262126 -7201.0852
1891900 298.84229 1.575551 -64596.658 420.08763 46.858053 51.931256 37.219733 -7202.2921
1892000 295.05817 1.5762017 -64572.601 81.052564 46.851605 51.924109 37.214611 -7175.0504
1892100 295.8364 1.5667148 -64574.74 -1461.6913 46.945981 52.028703 37.289575 -7183.7407
1892200 298.13618 1.5719642 -64569.86 -335.30798 46.893666 51.970724 37.248021 -7118.8356
1892300 295.5986 1.5734643 -64578.808 -191.82519 46.878758 51.954202 37.23618 -7080.6471
1892400 296.88934 1.5656079 -64578.23 -452.00234 46.957042 52.040962 37.298361 -7204.2618
1892500 297.06042 1.5736521 -64567.947 -821.85221 46.876893 51.952136 37.234698 -7090.1657
1892600 297.49728 1.571721 -64567.237 -639.91408 46.896084 51.973404 37.249942 -7145.6834
1892700 301.31144 1.5701851 -64568.389 -792.49956 46.91137 51.990345 37.262083 -7085.364
1892800 296.64601 1.5697093 -64559.839 457.05154 46.916109 51.995597 37.265848 -7061.0097
1892900 298.60305 1.5672082 -64550.36 -7.1049726 46.941054 52.023243 37.285662 -7135.3777
1893000 299.63557 1.5720568 -64551.872 620.96734 46.892745 51.969703 37.247289 -6923.9615
1893100 301.08666 1.5645561 -64561.162 -509.21868 46.967562 52.052621 37.306717 -7169.7051
1893200 297.76565 1.5688223 -64553.655 211.03077 46.924949 52.005394 37.272869 -7046.6749
1893300 297.68527 1.5671614 -64542.729 -89.166431 46.941521 52.023761 37.286033 -7119.5188
1893400 298.4174 1.5705333 -64539.353 498.78121 46.907903 51.986503 37.25933 -7141.6041
1893500 300.36475 1.5729131 -64543.97 531.49104 46.884234 51.960271 37.240529 -7153.2909
1893600 295.91741 1.5703697 -64544.742 -700.316 46.909531 51.988307 37.260623 -7132.1701
ERROR on proc 0: Out of range atoms - cannot compute PPPM (…/pppm.cpp:1891)
Last command: run 2000000
There may be an error with your dynamics, most likely the timestep is too large. Are you sure that you can use a 2 fs timestep with your potential ? What if you try with 0.5 fs ?
I’ll try that and get back to you. thanks.
Hi Simon,
I tried 0.5 fs . Lattice parameteres changed very smoothly, but same error happend:
2894400 298.47106 1.5609285 -65425.686 -604.63403 47.003918 52.092913 37.335595 -6876.2354
2894500 297.72643 1.5585531 -65429.184 1801.3231 47.027786 52.119365 37.354554 -6892.7499
2894600 298.67257 1.5553738 -65432.546 -386.07969 47.059807 52.154852 37.379988 -6904.0845
2894700 295.62551 1.5523568 -65433.885 -582.93046 47.090274 52.188619 37.404188 -6971.5315
2894800 298.50105 1.5507407 -65436.18 -895.09874 47.106626 52.206741 37.417177 -6987.3305
2894900 293.48321 1.5504095 -65437.342 26.045675 47.109981 52.210459 37.419842 -6959.8898
2895000 297.11028 1.5509814 -65440.74 -903.0242 47.104189 52.20404 37.415241 -6938.1794
ERROR on proc 4: Out of range atoms - cannot compute PPPM (…/pppm.cpp:1891)
Last command: run 2000000
MPI_ABORT was invoked on rank 4 in communicator MPI_COMM_WORLD
with errorcode 1.
I appreciate any comments and help on this.
Hi, it could be an overlap between your atoms (but it could be something else as well, you are not giving enough information here). Have you try minimizing the energy of the system?
Have you visualized your trajectory? When this kind of error happens after a substantial number of timesteps, it usually points to something unusual happening.
Another thing to look at would be the force field parameters. How did you obtain and assign them?
Yes, I have minimized the box and molecules in the lattice. But I think I need to simplify the problem and go back to the unit cell minimisation and equilibration.
Thanks so much, Alex. Yes, and something seriously wrong there. The force field is GAFF with CNDO partial charges. I have tested the force field parameters for the pure glycine and glycine/water systems. Those seem OK.
Since the system appears to be fully equilibrated, with stable energy and temperature fluctuations, and you were able to run for nearly 2 million time steps without incident, this is likely caused by a low-probability unfortunate event. This could be some combination of unphysically large forces from GAFF and too large a timestep. You could test the second part by making the tilmestep even smaller.
Are you saying that you didn’t use the (partial) charges that are part of the force field, but replaced them with charges your computed on your own?
Yes, I replaced partial charges with the CNDO but I got those from existing literature.
What kind of literature?
By replacing the charges you create a new force field and are disturbing the balance between LJ and Coulomb. Amber uses a particular charge fitting procedure for molecules and residues and if you don’t use that you may have too strong coulomb versus the LJ interaction which can lead to the problems you are seeing.
Thanks, Axel. I know what you mean.
I used the paper that examined the exact same system and conclude that GAFF with CNDO charges goes well: DOI: 10.1021/cg100906s
So I looked up this study and it appears that their results are such that in a system without solvent their (more polar) model is closer to experiment, but in solution worse than the conventional parameterizations.
Looking back at the information about your simulation the long “delay” here is worrying.
The long timestep of 2fs was also mentioned, but you tried a shorter one (but 0.5fs may not be short enough).
I would suggest to make some tests with shorter runs and check the the output for “dangerous builds”. You should check:
- 2fs timestep and fix shake for all bonds involving hydrogen atoms
- 0.25fs timestep and the same setup as before without fix shake
With 2fs timestep you will probably need a delay value of 0. But you can see what value you can use from the number of neighbor list builds versus the number of timesteps. Please keep in mind the law of diminishing returns and because of that, the biggest gain is from using delay 1 or delay 2 and growing this will reduce the gain. You can certainly a larger delay when running with a 0.25fs timestep and possibly also use every 2 instead of every 1 or even every 4. At any rate, there should be no dangerous builds except at the very beginning of the equilibration.
I sincerely appreciate your help, Axel. Will do that and get back soon.
Hi,
I run the following calculations on the optimized alpha and beta glycine:
equilibration run
neighbor 2.0 bin
neigh_modify every 1 delay 0 check yes
thermo_style custom step temp density etotal press cella cellb cellc evdwl
thermo 100
timestep 2.0
velocity all create 298 123987 dist uniform
dump trj_full all atom 1000 trj_full.lammpstrj
dump trj_nvt all atom 1000 trj_nvt.lammpstrj
fix fxnvt all nvt temp 298.0 298.0 2000 # iso 0.986923 0.986923 1000
run 1000000
unfix fxnvt
undump trj_nvt
dump trj_npt all atom 1000 trj_npt.lammpstrj
fix fxnpt all npt temp 298.0 298.0 2000.0 iso 0.986923 0.986923 1000.0
run 2000000
unfix fxnpt
fix fxnpt all npt temp 298.0 298.0 2000.0 aniso 0.986923 0.986923 1000.0
run 2000000
write_data data.post_npt nocoeff
lattice parameters and density seems fine for alpha glycine:
(a)exp=4.9669, (b)exp:11.4594, (c)exp:5.4231
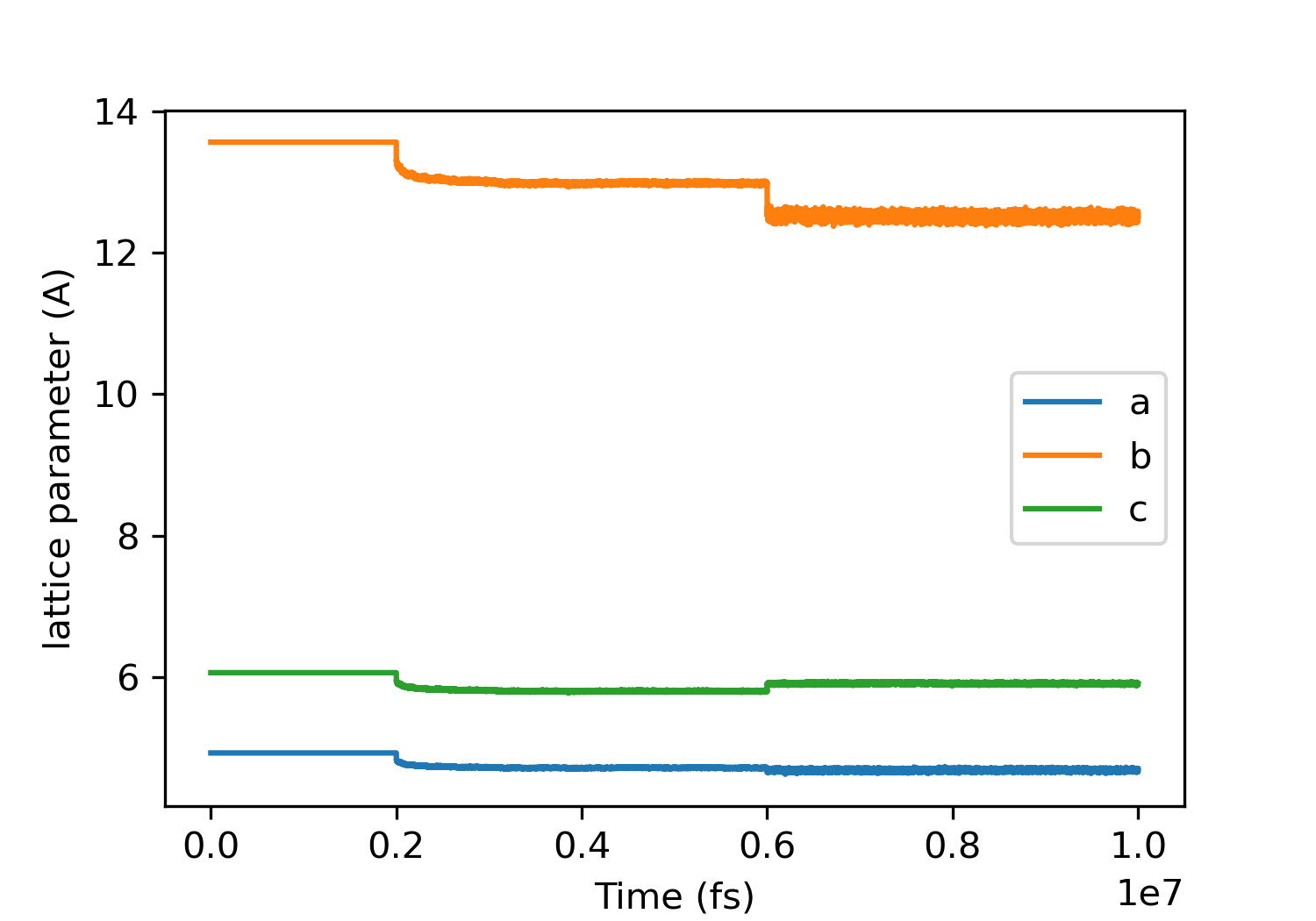
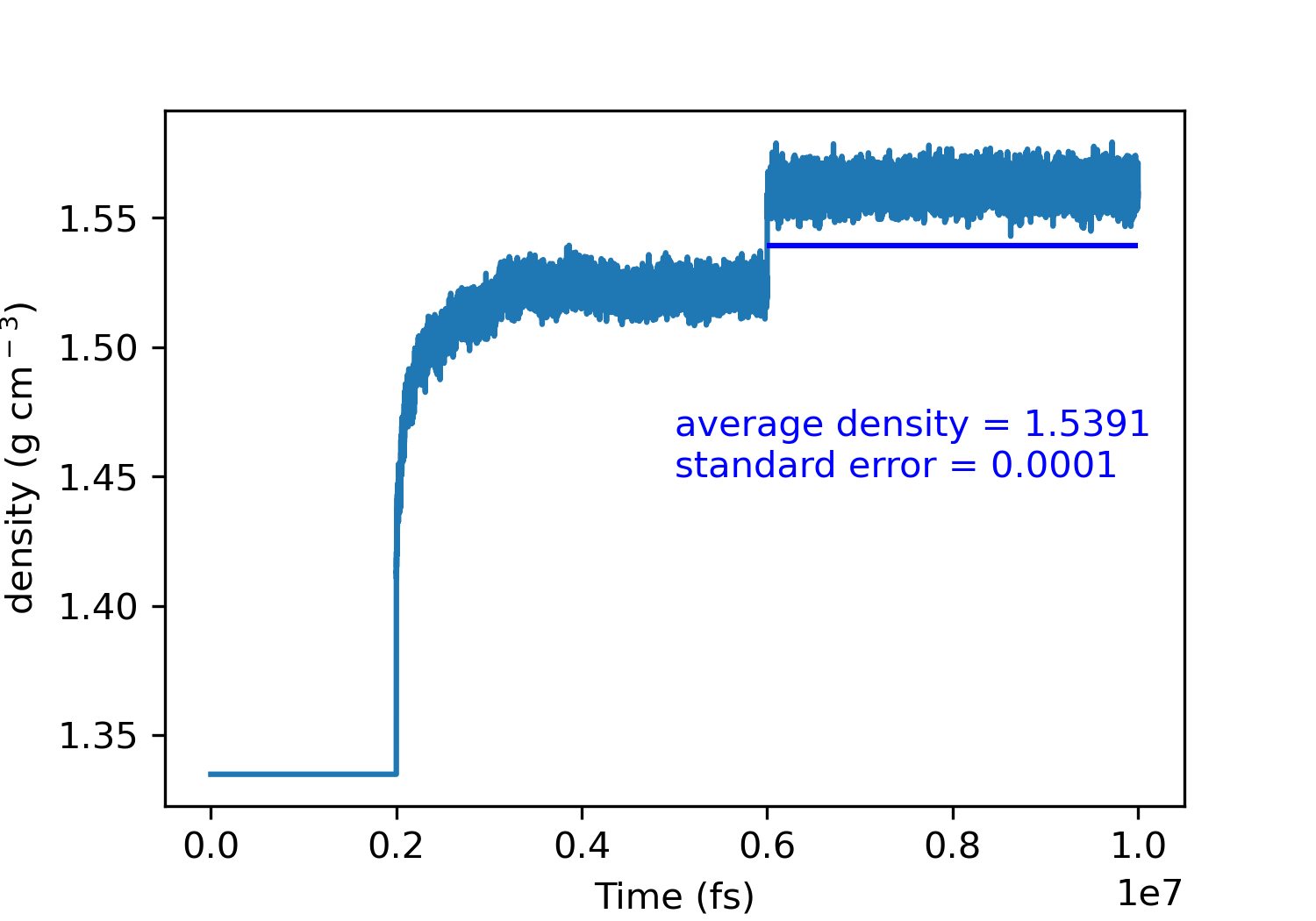
However, something strange happens for beta glycine with anisotropic NPT ensemble:
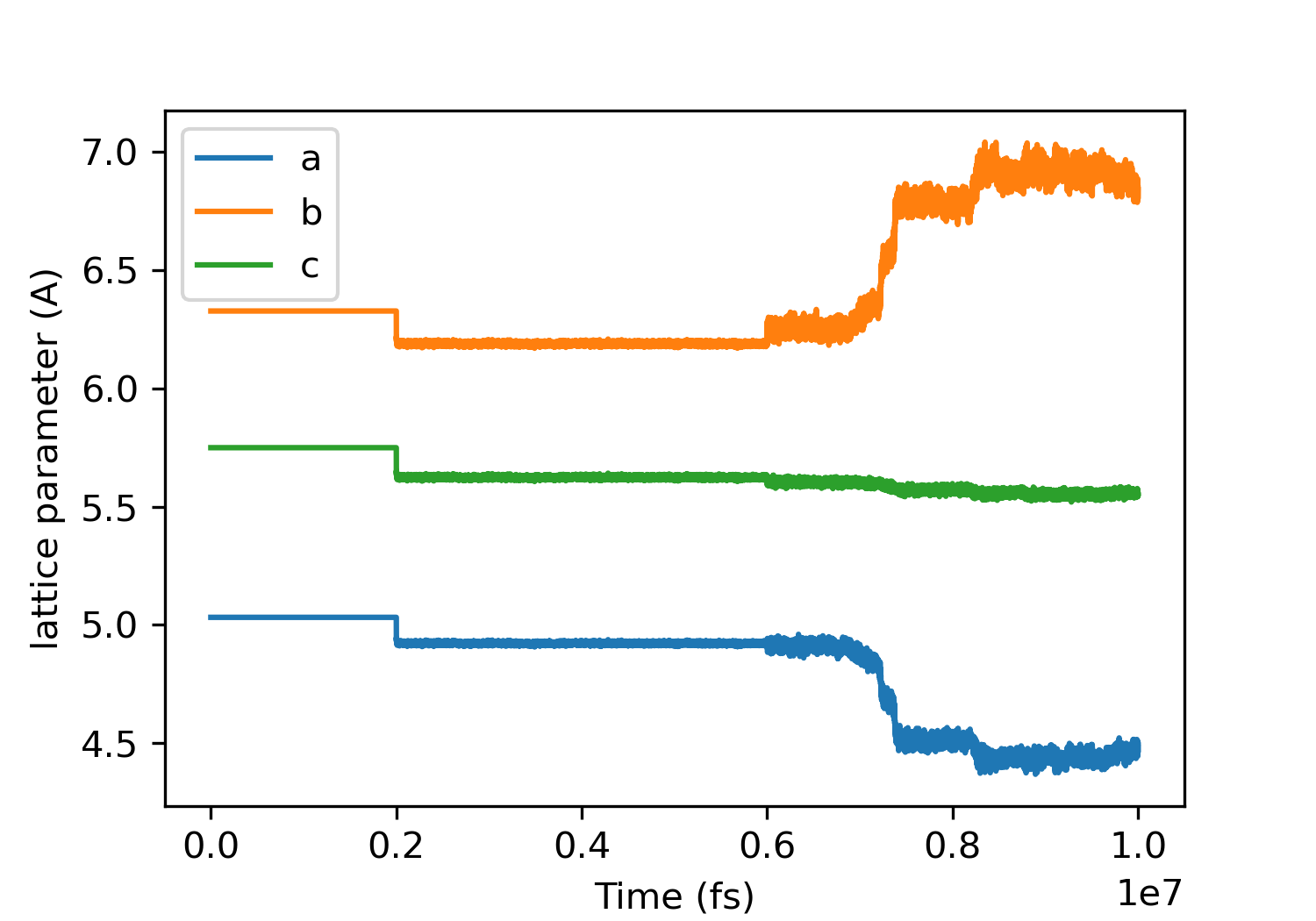
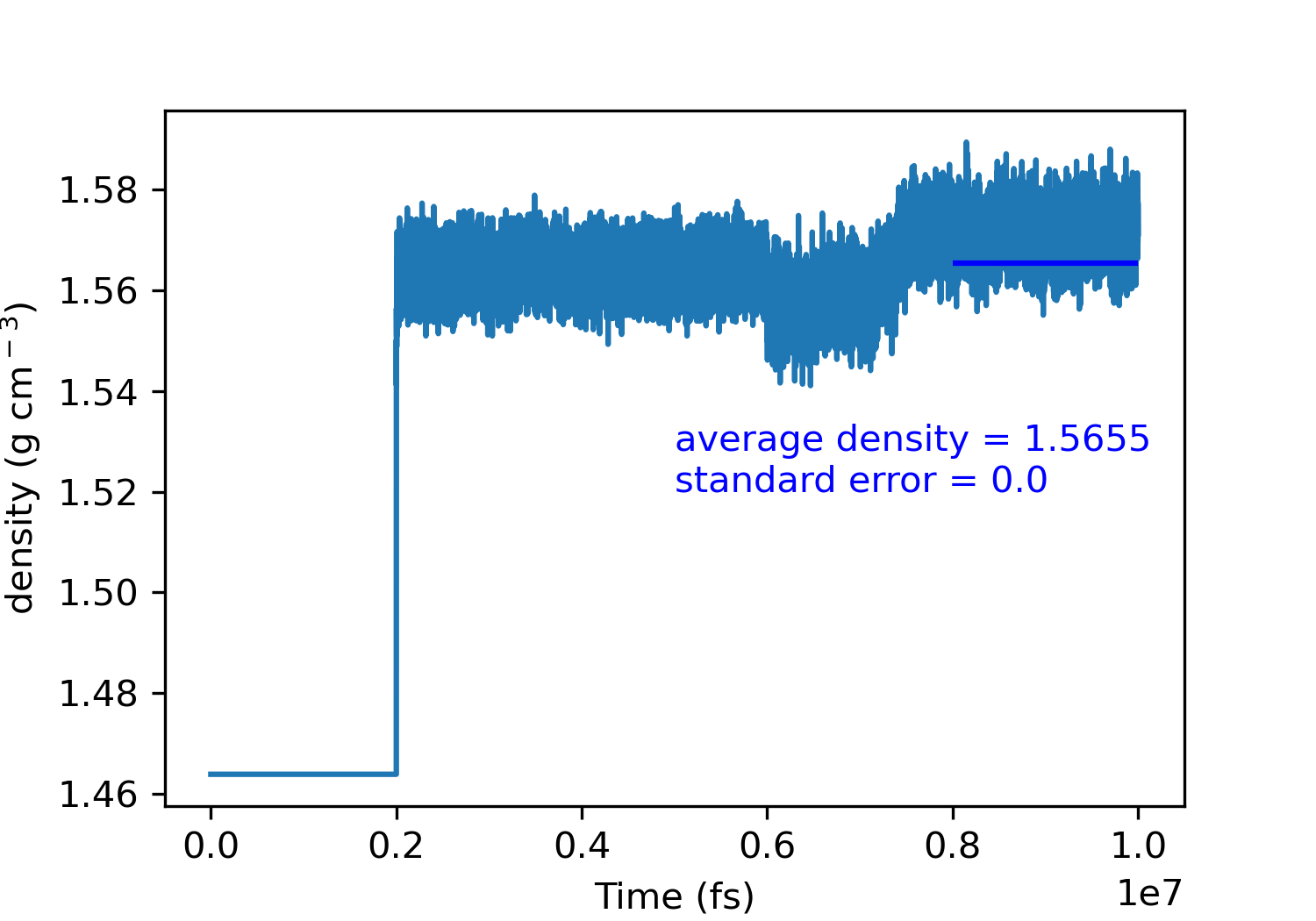
Fortunately, calculations didn’t crash and ‘delay’ command solved the problem.