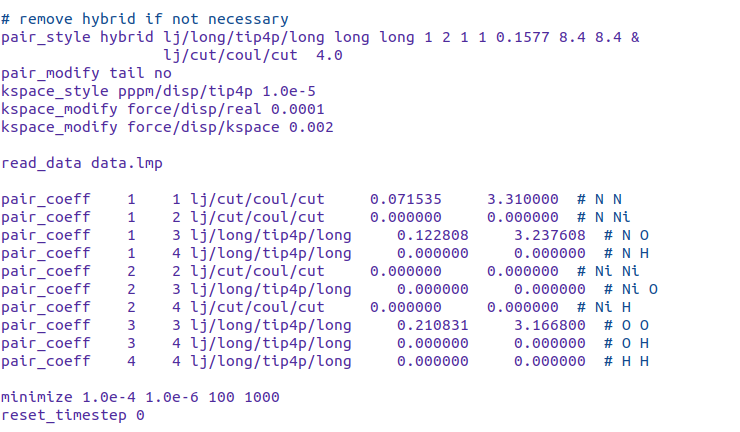
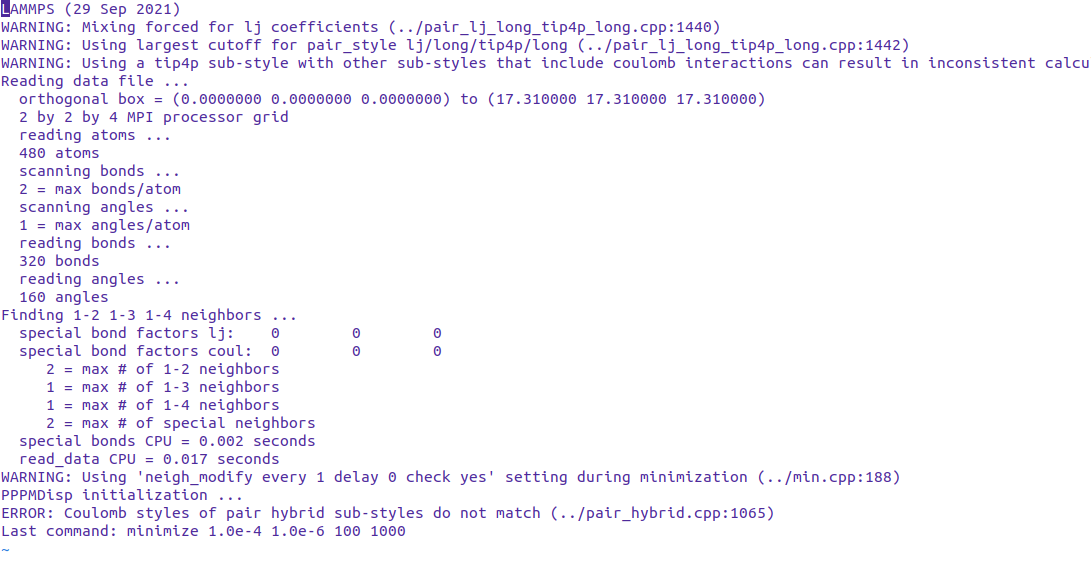
I have a system with 7 atom types, of which 5 are charged. I want to calculate only the short range coulombic interactions for some atom type pairs whereas full coulombic interactions for the remaining pairs. I tried using the coulombic pair styles as follows:
pair_style hybrid lj/long/tip4p/long long long 1 2 1 1 0.1577 8.4 8.4 &
lj/cut/coul/cut 4.0
I have used pair_coeff command to allocate the sub-styles to respective atom types.
Upon trying to run the simulation, I get the following error:
ERROR: Coulomb styles of pair hybrid sub-styles do not match (…/pair_hybrid.cpp:1065)
Last command: minimize 1.0e-4 1.0e-6 100 1000.
I am attaching the screenshot of my input file and the error for a better context. Please lend me a hand.
Thanks for quick response guys. But the simulation I am doing is for a specific purpose where I specifically need only the short range coulombic interactions. Thanks for the help again.
You can either have only all coulomb short ranged or all coulomb long-ranged.
The K-space style does not know which is which and cannot separate out individual pairs (it works by computing the long-range coulomb interactions of each atom with all other atoms and their periodic replica at the same time. So mixing would double count interactions.
But it already makes no sense at the conceptual level. Coulomb interactions are coulomb interactions and the short-range or long-range styles are merely different ways to compute the same with different levels of error. Thus, if you feel you have the need to mix those, you likely have some conceptual problem with the kind of model you are trying to set up.
Sorry that I did not see your later reply due to some unavoidable circumstances. Thanks for the elaboration. Let me give a little perspective as to what I meant by specific purpose:
I have a crystal structure and their are some foreign (guest) particles in it. I want to calculate the interaction of these guests with the nearest (within a certain cutoff) guests, but not with all the guests in the system. The interaction with the remaining guests is being computed in a separate simulation and being added. The need to separate out the near and far interaction arises from the statistical mechanics derivation for the partition function of my system, which is the basis for the model of my system.
I hope this explains what I wanted to say earlier.
You can straightforwardly proceed if you set your “guest” particle’s charge to zero, and use hybrid/overlay with a pair table style to impose whatever short-ranged forces and energies you like.
But that is still very shaky theory. Electrostatic energies are not linear in the superposition of charge densities. This shows itself operationally in the simple question of what your simulations even mean with a net non-zero charge (as it would be if you followed the pair table suggestion). Yes, Ewald summation is often presented as a charge density repartitioning to decompose short and long-ranged forces, but the energy is not simply calculated as the sum of “short- and long-ranged energies” – it has to be carefully accounted for to reconstruct the correct, desired energy.
I would recommend running your simulations with straightforward electrostatics and then, if you want to fit a theory to it, using post-processing to partition the energies you have observed (for example using compute group/group).