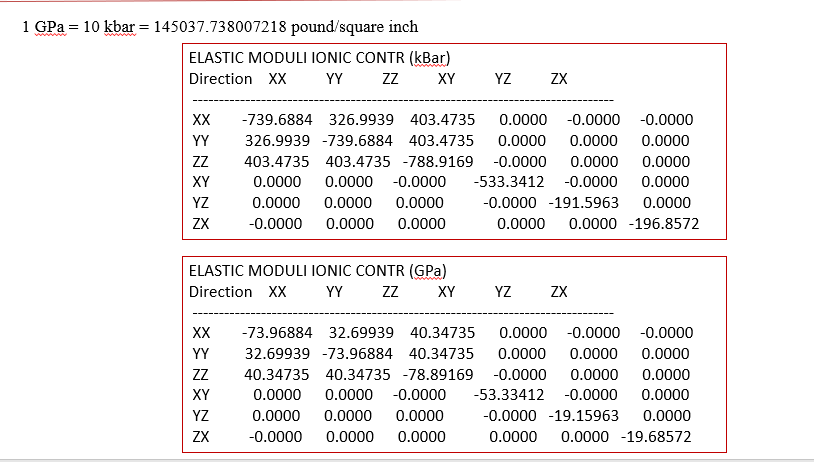
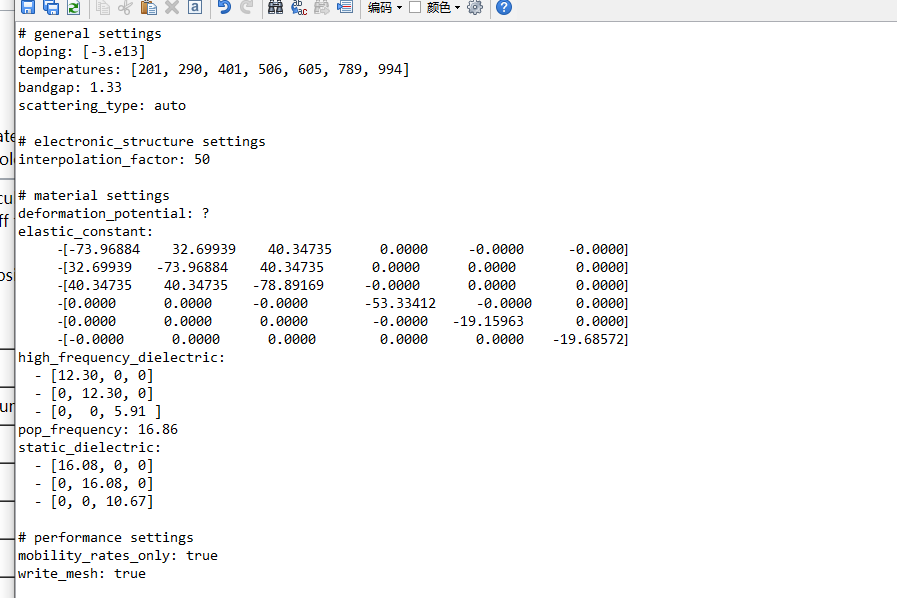
i Alex,
Recently I met with a problem when checking the output of deformation potential tensor calculation and the output of amset run. I found that the band indice of VBM and CBM in output of deformation calculation are always one number smaller than those in the output of amset run.
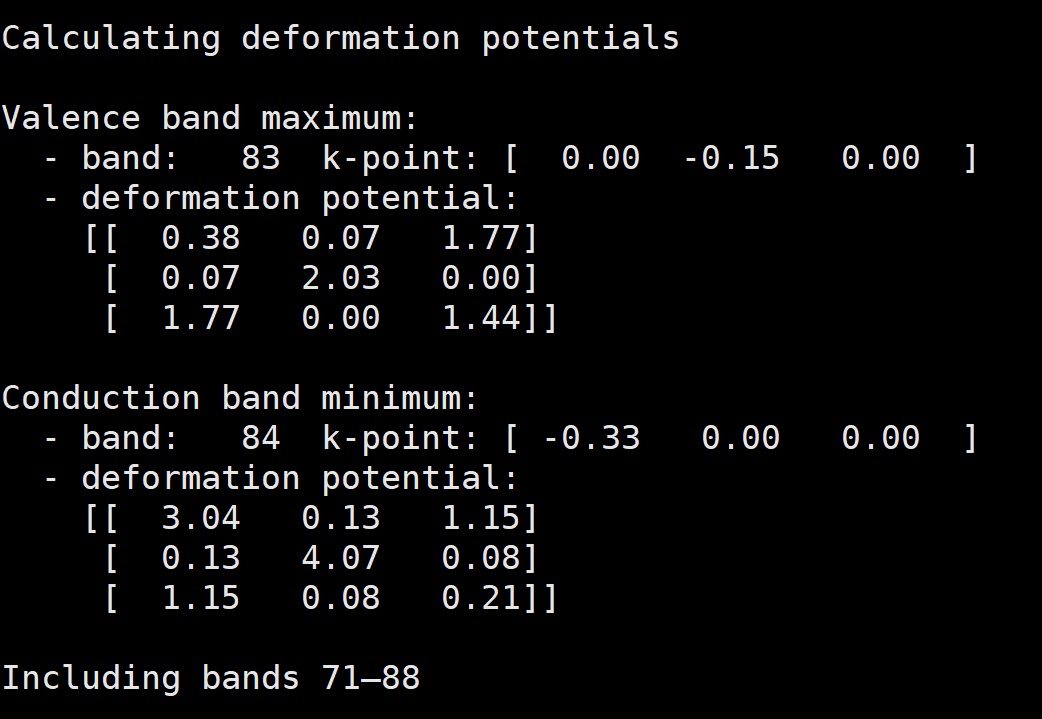
As you can see, in my deformation output, the VBM is #83 and CBM is #84
Valence band maximum:
- band: 83 k-point: [ 0.00 -0.19 -0.43 ]
- deformation potential:
[[ 0.70 0.03 4.88]
[ 0.03 1.76 0.07]
[ 4.88 0.07 2.39]]
Conduction band minimum:
- band: 84 k-point: [ 0.50 0.00 0.00 ]
- deformation potential:
[[ 3.52 0.07 6.50]
[ 0.07 3.50 0.07]
[ 6.50 0.07 0.28]]
Including bands 69—88
but in amset run output, the VBM is #84 and CBM is #85
Input band structure information:
├── # bands: 100
├── # k-points: 602
├── Fermi level: 4.942 eV
├── spin polarized: False
└── metallic: False
Band gap:
├── indirect band gap: 0.723 eV
├── direct band gap: 1.029 eV
└── direct k-point: [0.17, 0.00, -0.00]
Valence band maximum:
├── energy: 4.561 eV
├── k-point: [0.00, 0.19, 0.43]
└── band indices: 84
Conduction band minimum:
├── energy: 5.284 eV
├── k-point: [0.50, 0.00, -0.00]
└── band indices: 85
~~~~~~~~~~~~~~~~~~~~~~~~~~ INTERPOLATION ~~~~~~~~~~~~~~~~~~~~~~~~~~
Getting band interpolation coefficients
└── time: 2.5427 s
Interpolation parameters:
├── k-point mesh: 33x71x19
└── energy cutoff: 1.5 eV
Interpolating spin-up bands 69-88
└── time: 0.4054 s
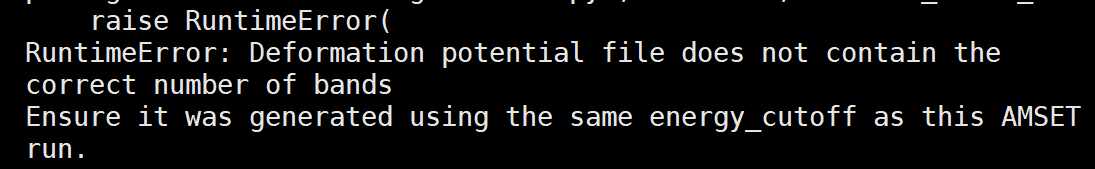
And in a most serious case, the total number of bands included in interpolation varies in deformation.h5 and wavefunction.h5, which leads to the breakdown of amset run.



I can guarentee that I used exactly the same INCAR (except two tags: LORBIT and ICORELEVEL)and KPOINTS in deformation calculation and wavefunction calculation. Why is this error happening? Looking forward to your reply.
Thanks,
Zhi