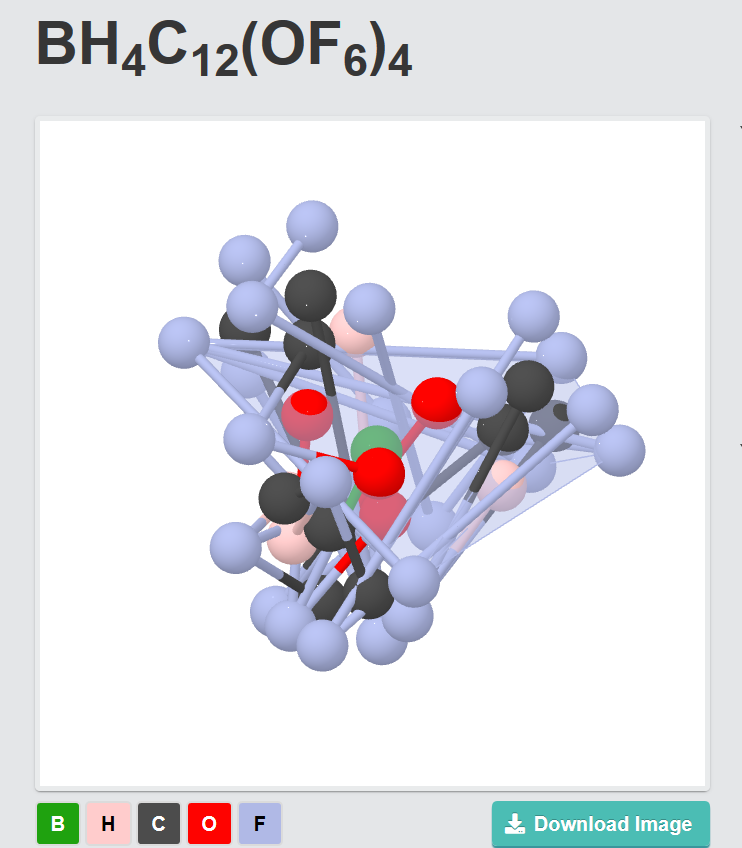
Hello
I have tried to upload a .cif-File. Only an error message appeared:
“Could not parse uploaded file. If this seems like a bug, please report it. Crystal Toolkit understands all crystal structure file types and molecule file types supported by pymatgen.”
Thanks @Leonie, I’ll look into it. Just taking a quick glance I suspect the issue is that these CIF files do not contain any information about the crystal lattice, so may be out of spec. Whether in spec or not, this would be the reason pymatgen is failing to parse them. In general, CIF files are meant to store crystallographic information (the ‘C’ in CIF), and I haven’t seen them storing just isolated molecules previously. It could be that this is completely valid and we just need to fix the pymatgen implementation. You could also try exporting as .xyz
Thanks for the answer!
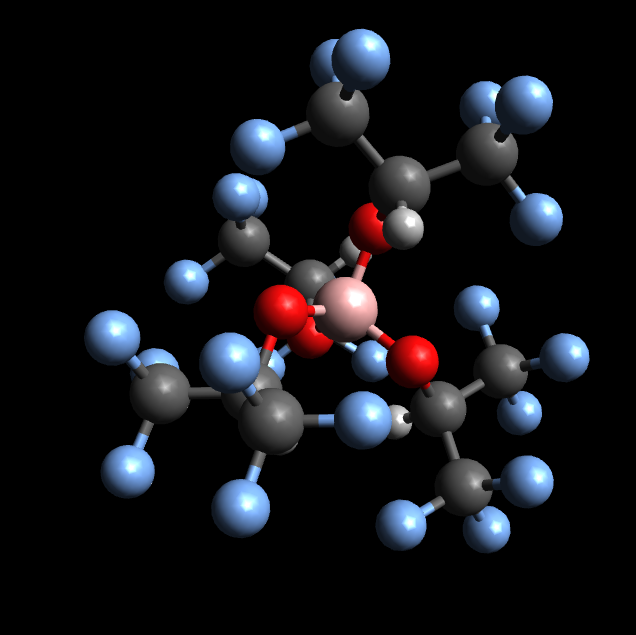
I’ve tried to upload the .xyz file. It worked but the preview of the material looks not identical to the created file.
The file was original created with Avogadro and exported from a cml-file to a .xyz-file.
The difference is due to a bug in the bonding algorithm for molecules (which has been fixed but not deployed to the website yet).
Note that the atomic positions are correct and the same between both. The xyz file does not contain information on which atoms are bonded to which other atoms, so we have to work that out. There are many algorithms in pymatgen which try to answer the question of if two atoms are bonded. Unfortunately, the ones for molecules had a bug where atom labels were permuted leading to the wacky bonds shown. As mentioned, this has been fixed in pymatgen, and will be fixed on the next update to the website too.