Dear LAMMPS users,
I’m trying to simulate a polymer model which is based on this paper. In this paper the authors have taken
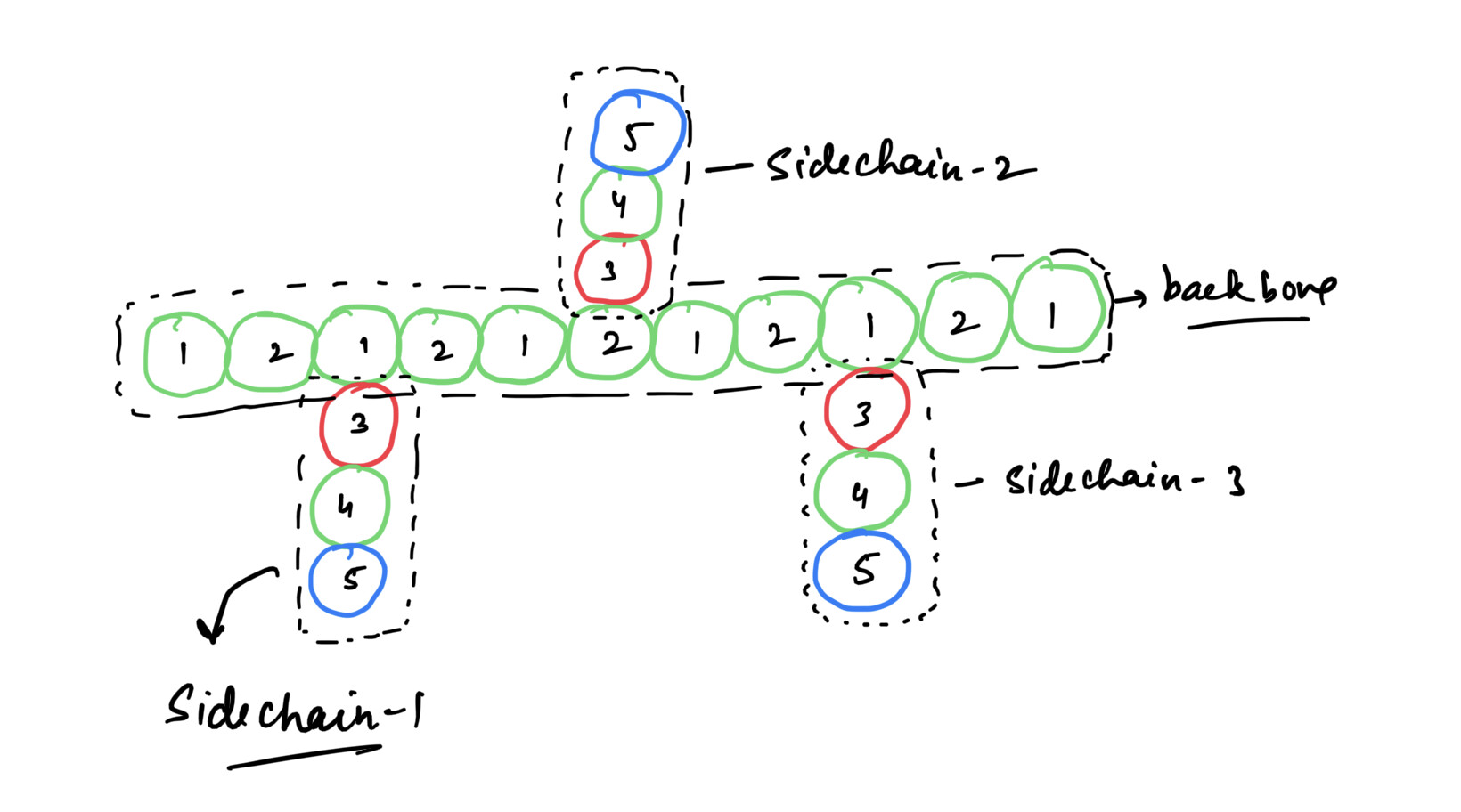
a polyMPC which consists of an aliphatic backbone with side chains at regular intervals as shown in below figure.
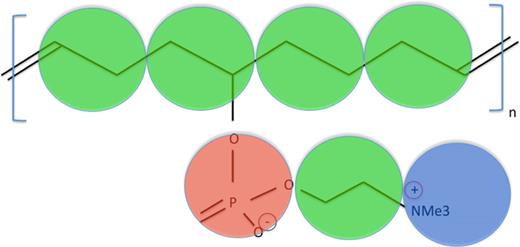
Each ethyl group is represented as a coarse grained bead. These are connected by harmonic springs to represent the linear backbone of the chain. Each side group is taken as a rigid rod consisting of three beads, anionic phosphate group, and cationic NMe+3 group at both ends of the rod connected by a hydrophobic ethyl group in the middle. The anion is connected to the backbone by a harmonic spring.
Further they also have mentioned,
To avoid any interference of LJ interaction on the harmonic bond potential, the LJ interaction between the adjacent beads along the backbone and also the LJ interaction between the backbone hydrophobic bead and the attached anion were turned off.
and lastly,
Finally the interaction between the beads within a side chain (rigid body) is turned off for computational efficiency. The beads of side chain interacts with the beads of backbone as well as the beads of other side chains but they do not interact with other beads in the same side chain.
With this background, I have a question regarding implementation of this in LAMMPS. I made such a scheme,
Say, I take 5 types of atoms. 1 & 2 belonging to the backbone whereas 3,4,5 belonging to the side chain. Since the interactions between beads within same sidechains are to be turned off. I excluded 3-4 & 4-5 interactions using neigh_modify but this will prevent interaction of atom type 3 from sidechain-1 to say atom type 4 from sidechain-2. So clearly, this will not work. If I assign every bead in all the sidechain a different type and then group the three beads in one group as, say, sc-1 and repeat the process for all other sidechains but again LAMMPS has an upper bound on both the number of atoms and number of groups and since I want to make this system for N=175 (N being number of repeat units) again this will not work. I thought of taking side chains atom type 3, 4 & 5 and group the atoms basing on atom ids and then using neigh_modify again this has the same number of group issue. I believe treating them rigidbody will be achieved via fix rigid/nve/small command. I havent implemented it yet. I got stuck with treating them non-rigid in the first place so I didn’t make much efforts.
How do i implement the above mentioned protocol in LAMMPS? I tried mailing the authors but its been a while since I have received a response from them.
Thanks and Regards
Akshay
This not how you handle this. Instead you should look into the special_bonds command. If I read the description correctly, you should be using special_bonds lj/coul 0 1 1. This will exclude all LJ interactions that have a bond between them. No neigh_modify commands needed.
These are called “exclusions” and you should read up on this and in general how molecular force fields are designed and implemented in your favorite text book on MD simulations.
1 Like
Dear Axel,
Thank you so much for quick response and definitely I’ll read more about ‘exclusions’.
One more thing, The authors of this paper have treated all the sidechains as rigidbody.
The beads belonging to the side chains are treated as rigid bodies. The velocities and positions of the beads belonging to the side chains are updated by the rigid body algorithm adopted from Miller et al. in LAMMPS. The velocity and the position of the beads belonging to the backbone of the polymer chain are updated by the velocity-Verlet algorithm.
I put a quick test simulation where I made a smaller chain and with 10 sidechains and type 3, 4 & 5 were respective atom types for each bead in the side chain. Further, I grouped them as:
group three type 3
group four type 4
group five type 5
group s1 union three four
group sidechain union s1 five
After this I did:
fix myrig sidechain rigid/nve group 3 three four five langevin 1.0 1.0 100 123456
this worked as in the simulation ran somehow but the outcome was extremely unphysical. What was happening was all the type 3 beads were moving in sync together, and same with type 4 and 5.
I looked into the documentation it said
The rigid/small styles are typically best for a system with a large number of small rigid bodies.
I wanted to ask, am I approaching the problem in a wrong manner? Does this system qualifies as a system containing “large number of small rigid bodies”? How large is large enough so that one uses this rigid/small or rigid/nve/small command. Should I be using three different fixes for each atom type in the sidechain? I actually tried different fixes as well but the outcome wasn’t much different from the earlier one, still unphysical.
Thanks and Regards
Akshay
That is what you requested in your input, but that is not likely what the authors of the paper did. They obviously created individual rigid bodies from the side chains. This is not easily done with groups and likely you will run out of groups (there is a limit). Instead, you should be using the “molecule” keyword to group rigid bodies by molecule ID.
Large enough is when fix rigid/nve/small is faster than fix rigid/nve, which will be most visible when running a larger system in parallel with MPI. The biggest limitation of fix rigid and the other non-small variants is that the handling of the rigid bodies is global and thus does not parallelize well, whereas with the “/small” the rigid body information is stored with an atom of the the rigid body and the rigid bodies are handled in a distributed way in parallel. That adds some overhead but parallelizes much better.
1 Like
Dear Axel,
Correct if I’m wrong but won’t we still need to make the groups?
fix ID **group-ID** style bodystyle args keyword values ...
I mean even if the bodystyle is molecule still one needs to input a group ID of the respective species in the system on which this fix will be applicable. Say, I’ve given different molecule ID to each sidechain I believe that I still need to form groups based on molecule IDs.
No. Please study the documentation and the micelle example.
1 Like