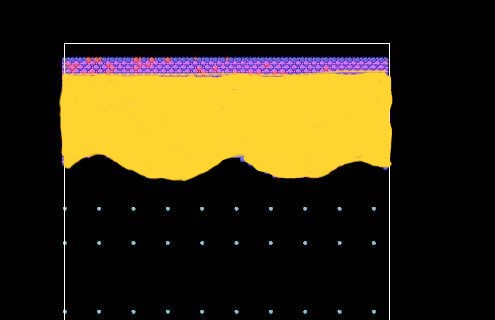
I am currently simulating the condensation of Argon atoms on a Sine - nanostructured substrate. I need to define the curved region on the surface accurately to get accurate results.
I attempted to start this approach using LAMMPS, defining a variable ‘v’ to represent the sinusoidal surface. Only the create_atoms command works with this approach. As the region command in LAMMPS does not support defining surfaces with a variable(only standard shapes like prisms, spheres, and rectangular blocks), I am facing challenges in accurately defining our surface.
The file “rough_2.txt” is the substrate that I had created beforehand, I was just using the create_atoms command to check if the variable ‘v’ is defined properly.
# Initialize Simulation Parameters
package omp 18
units metal
dimension 3
boundary p f p
atom_style atomic
neighbor 1 bin
neigh_modify delay 3
# Read Initial Atomic Configuration
read_data rough_2.txt
# Define regions for the simulation box
region wall1 block 0 160 0 15 0 80 units box # Solid region
region liqA block 0 160 17.42 57.42 0 80 units box # Liquid region
region gasA block 0 160 60.32 930 0 80 units box # Gas region
region gas block 0 160 65 925 0 80 units box # Another gas region
region liqB2 block 0 160 938 972 0 80 units box # Another liquid region
# Create a solid upper wall region (before cutting)
region upper_wall block 0 160 985 1042 0 80.36 units box
variable xx internal 0.0
variable yy internal 0.0
variable v equal "(v_yy - 1029 +39 - 6*sin(1.5*0.06238*v_xx)) > 0.0"
# Continue with the rest of the simulation
lattice fcc 5.79
create_atoms 3 region liqA
lattice fcc 34
create_atoms 3 region gasA
create_atoms 3 region liqB2
lattice fcc 3.92
create_atoms 4 region wall1
lattice fcc 3.92
create_atoms 4 region upper_wall var v set x xx set y yy
write_data whole_new.dat
The files:
rough_2.txt (1.2 MB)
Has anyone encountered a similar issue or found a workaround to define complex surfaces like sinusoidal geometries in LAMMPS? Any advice or alternative approaches would be greatly appreciated.