Hello LAMMPS Developers and Users @julient,
I am running a spin-lattice coupling simulation in LAMMPS using the pair_style hybrid/overlay command with four different potentials: pair_style hybrid/overlay eam/fs spin/exchange 4.5 spin/neel 4.5 spin/dmi 4.5, and the contents of in.Fe.lmp file are as follows
# LAMMPS Input file for spin-lattice dynamics of bcc iron (Fe)
clear
units metal
dimension 3
boundary p p p
atom_style spin
atom_modify map array
#lattice structre
lattice bcc 2.855
region box1 block -50 50 -50 50 -30 30
region cyl_region cylinder z 0 0 15 -3 3
create_box 1 box1
create_atoms 1 region cyl_region
mass 1 55.845
group Fe type 1
pair_style hybrid/overlay eam/fs spin/exchange 4.5 spin/dmi 4.5 spin/neel 4.5
pair_coeff * * eam/fs Fe_mm.eam.fs Fe
pair_coeff * * spin/exchange exchange 4.5 0.025498 0.281 1.999
pair_coeff * * spin/dmi dmi 4.5 0.005498 0.0 0.0 1.0
pair_coeff * * spin/neel neel 4.5 0.005498 0.281 1.999 0.0 0.0 1.0
# neighbor
neighbor 0.1 bin
neigh_modify every 10 check yes delay 20
## initial condition setting
set group all spin/atom/random 1235489 2.2
#set group all spin/atom 1.0 1.0 0.0 0.0
velocity all create 1 4928459 rot yes dist gaussian
timestep 0.0001
fix 3 all nve/spin lattice moving
thermo 100
thermo_style custom step temp etotal
run 2000
Issue Description
When I run my simulation with all four potentials included, the execution does not proceed beyond the initial setup stage. The output remains stuck at:
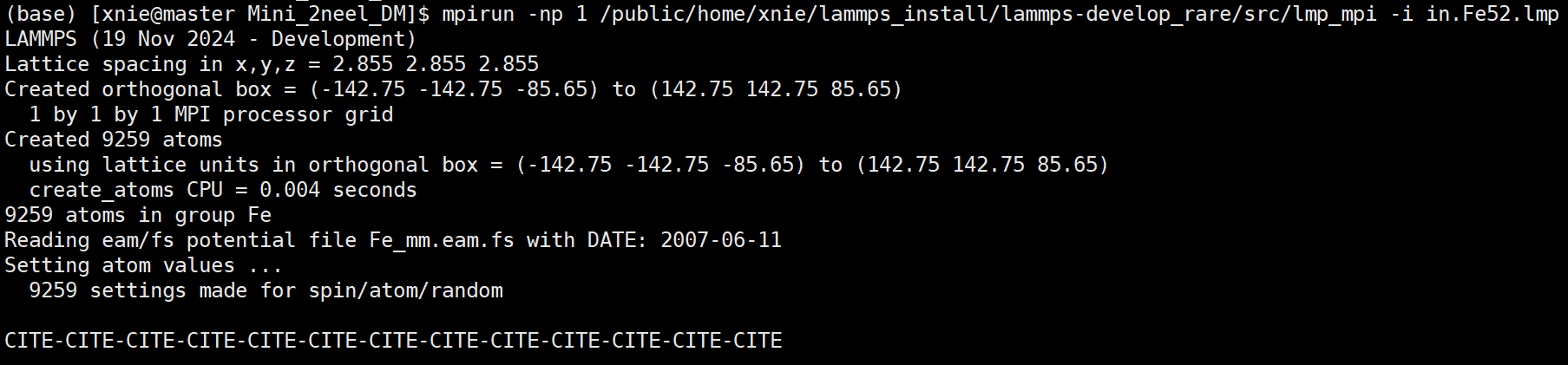
The simulation does not crash or give an explicit error; it just does not proceed beyond this point. LAMMPS Version is LAMMPS (19 Nov 2024 - Development), which is compiled with support for spin-lattice coupling.
What I Have Tried
- Reducing system size: The issue persists even with a much smaller system of 63 atoms.
- Single-core vs. multi-core: The problem occurs with both “mpirun -np 1” and “mpirun -np 24” , so it does not appear to be MPI-related.
- Removing one potential at a time:
- Any combination of three potentials works fine.
- The issue only appears when all four potentials (“eam/fs” , “spin/exchange” , “spin/neel” ,
spin/dmi) are used together.
4.Reducing interaction cutoff: Changing the cutoffs for “spin/neel” , “spin/dmi” , and “spin/exchange” did not resolve the issue.
- Replacing “spin/dmi” or “spin/neel” with another potential: If I replace one of them with another interaction (e.g., biquadratic exchange), the simulation runs fine, which suggests a conflict between
spin/neelandspin/dmiwhen used together.
Questions for Developers
Is there a known limitation or conflict when using “spin/neel” and “spin/dmi”together with** “eam/fs” and “spin/exchange” under “hybrid/overlay”? My research has to require the use of all four potential at the same time, how should I solve this problem?
Any insights or suggestions would be greatly appreciated!
Thank you in advance!