Hi,
I’m simulating a gaseous system involving 16 atoms. I did a NVT simulation at temperature 420K and a pressure of 0.98 atm for 5 nanoseconds.
timestep 2.0
variable dt equal 2.0
variable ext_temp equal 420
variable Tdamp equal 100.0*${dt}
fix 2 all nvt temp ${ext_temp} ${ext_temp} ${Tdamp}
###
write_restart ./restart_file
run 5000000
write_restart ./restart_file # just in case
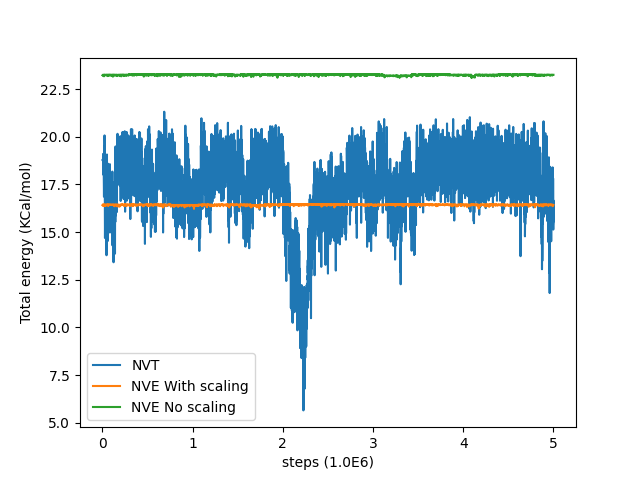
During this the total energy reached an equilibrium around 17.5 KCal/mol.
Following this, I wrote the final atomic positions and velocities in a ‘restart_file’ and I use this to start a new NVE simulation.
read_restart restart_file
fix 2 all nve
However, I notice that in the new NVE simulation the temperature suddenly jumped to 520 K and the total energy jumped to 23 KCal/mol. Why is this sudden jump happening? I again ran another NVE simulation in which I scaled the velocities using
velocity all scale 420
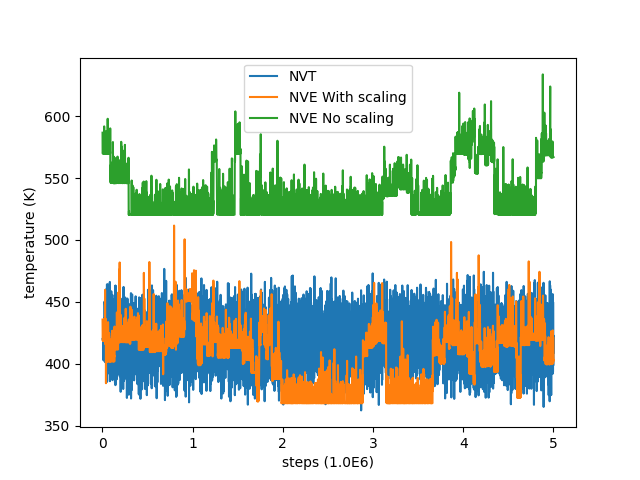
and in this case the temperature and total energies were more close to the average values in the previous NVT simulation. I’m attaching with this two plots showing the fluctuations in temperature and energy for the NVT simulation, the NVE simulation without velocity scaling and with velocity scaling.
As we can see the temperature in the NVT simulation never goes above 470K and the total energy never goes above 21 KCal/mol, so this issue could not have been caused by the restart file being at a higher temperature point during the writing. Also the final value of temperature and total energy in the NVT simulation is 400K and 15.5 KCal/mol respectively.
Then why is this sudden rise happening?
Without access to the data file and the potential table files, it is difficult to try to investigate since it is not possible to reproduce what you see.
Please also note, that a system with just 16 atoms is ultra small and thus all properties that get averaged over all atoms will have very large fluctuations.
InputData.zip (125.9 KB)
Hi, here are the input files for re-creating the simulation. I’m actually using this 16 atom setup as an example to learn LAMMPS. I understand that temperature fluctuations are normal and expected for such a small system. But here I observe a sudden jump from 420 to 520 immediately after reading the old velocities from the restart file, this made me suspicious that there is something wrong with the reading of velocities from the restart file.
I cannot see this. To confirm the restart is working as expected I am adding the line:
write_dump all custom restart.dump id type x y z vx vy vz modify sort id
after writing the (final) restart file in the NVT run and then before starting the run in the NVE input and I get identical files. Also the thermo output continues exactly where it ended (I reduced the number of MD steps to 5000 to speed things up).
Per MPI rank memory allocation (min/avg/max) = 6.051 | 6.051 | 6.051 Mbytes
Step Temp Density E_pair Press
0 420 0.0038566216 -2.3555202 0.97261756
1000 397.04158 0.0038566216 -2.8399809 0.73529981
2000 479.8922 0.0038566216 -1.5425 0.90795311
3000 467.07691 0.0038566216 -1.9488045 0.88597543
4000 419.45308 0.0038566216 -2.3136951 1.0227926
5000 394.94252 0.0038566216 -1.3149848 0.72531211
Loop time of 0.00344005 on 1 procs for 5000 steps with 16 atoms
and
Per MPI rank memory allocation (min/avg/max) = 6.051 | 6.051 | 6.051 Mbytes
Step Temp Density E_pair Press
0 394.94252 0.0038566216 -1.3149848 0.72531211
1000 416.06572 0.0038566216 -2.2597441 1.1059007
2000 397.41488 0.0038566216 -1.425531 0.72327556
3000 413.08765 0.0038566216 -2.1449398 0.78592875
4000 424.69679 0.0038566216 -2.7012437 0.76074897
5000 405.24956 0.0038566216 -1.8131032 0.71891445
Loop time of 0.00359181 on 1 procs for 5000 steps with 16 atoms
Thank you and also for your time to analyze this. So what could be the reason for this variation in the average total energy of these two simulations? Should I repeat the same calculation with more number of atoms? My doubt is:
why the average energy obtained when I directly read the velocity from the NVT simulation at 420 K is different from the average energy I get when I scale the velocity to 420K ?
These are all topics for a discussion with your adviser. This is about the science and not about LAMMPS.
This is because of equilibration and fluctuations. The quantity that should be conserved is the total energy. However, with finite size time steps and numerical integration, there are some fluctuations to be expected. Especially for very small systems, the energy fluctuations on both kinetic and potential energy can be significant. If you rescale the velocities, you are changing the balance between kinetic and potential energy.
The change when switching from NVT to NVE may be due to the small system size where the number of degrees of freedom in the Nose-Hoover chain fictitious oscillators matter relative to the total degrees of freedom.
At any rate, this topic is resolved. There is no problem reading velocities from a restart file.
Then the discrepancy is very straightforward, to me:
You have a very small system (16 atoms!! Even Rahman’s 1960s papers had more atoms than that!!) in gas phase that you have thermostatted with Nose-Hoover (so you are artificially correlating all the atoms’ velocity).
Thus it is very possible for the total energy to fluctuate significantly step by step under NVT integration.
fix ave/time in your script is taking an average over 50 snapshots spaced 20 steps apart (or vice versa, I don’t remember). There is no guarantee that that average is the same as the instantaneous energy of the single configuration recorded in the restart file.
You just happened to record a restart when the instantaneous energy was quite different from the 1000-step / 50-snapshot average.
On the other hand, thermo output records the instantaneous values of the recorded variables. So if the thermo output had discrepancy between writing the restart and loading from the restart, that would genuinely be grounds for concern.