Hi,
I’m trying to simulate a 16-atom MD system in LAMMPS using a tabulated potential.
The tabular potential I use in this example is actually obtained from LAMMPS itself using the pair_write command.
I first use the built-in Lennard-Jones (LJ) potential with these parameters:
pair_style lj/cut 10.0 # LJ with 10.0 Å cutoff
pair_modify tail yes # Analytic long-range tail corrections
pair_coeff * * 0.0715 3.31
pair_write 1 1 1600 r 1.0 13.9 table_pot1.txt mypot
Then, I read back this same tabulated potential for the simulation.
However, when I run the simulation, I get the following error:
15 atoms in group xenon
1 atoms in group rubiduim
Generated 0 of 1 mixed pair_coeff terms from geometric mixing rule
Setting up Verlet run ...
Unit style : real
Current step : 0
Time step : 2
ERROR on proc 0: Pair distance < table inner cutoff: ijtype 1 1 dist 0.772383999
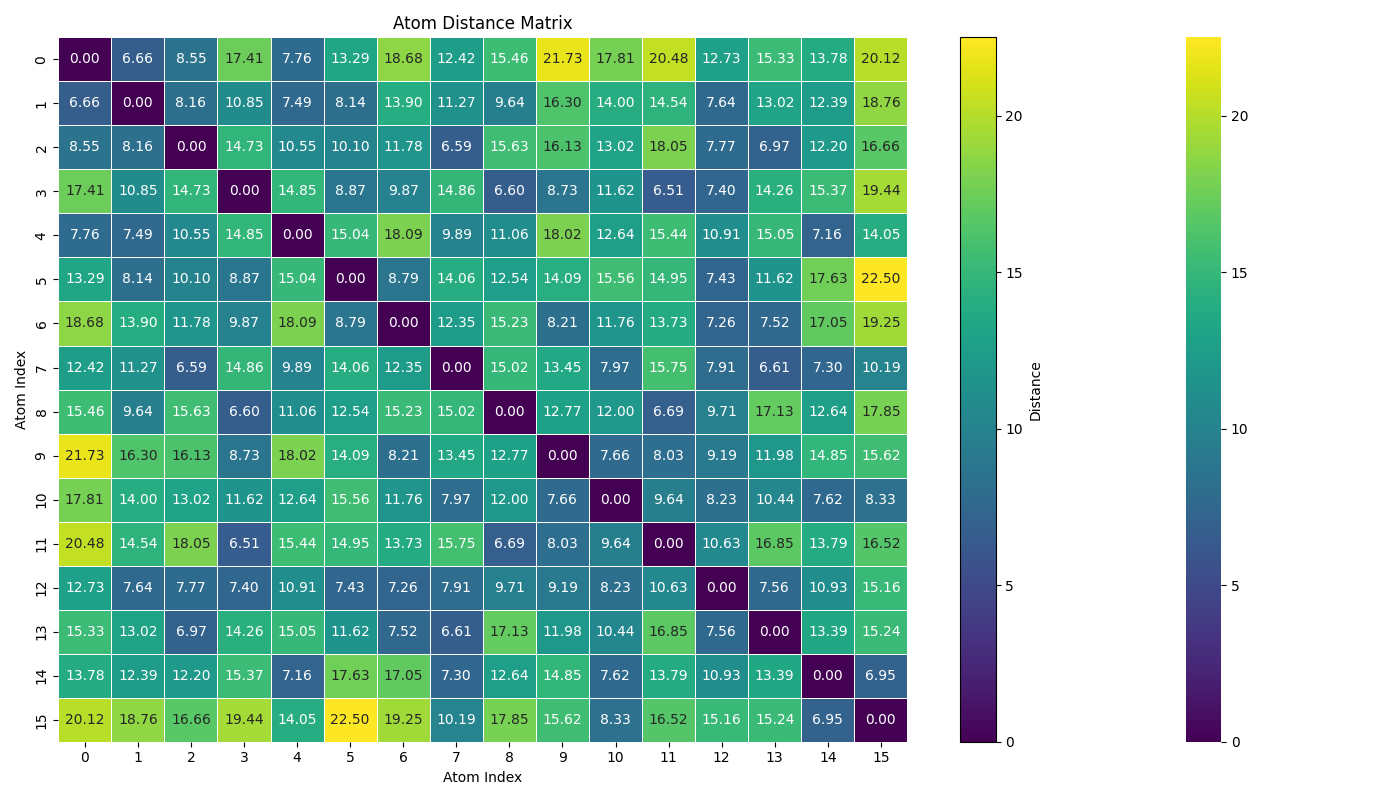
This suggests that two Xenon atoms are 0.77 Å apart, which is unphysical.
But when I manually check the distance using a Python script, the minimum actual distance is 6.51 Å. The simulation runs fine if I use the built-in LJ potential, but it fails when I read in the same potential as a tabulated file.
Here is my LAMMPS input script: LAMMPS_INPUT_FILE - Pastebin.com
Here is the atomic configuration File (Rb1Xe15.cfg): Rb1Xe15.cfg - Pastebin.com
What I Have Tried
Manually checked interatomic distances:
- Used a Python script to compute pairwise distances.
- Minimum distance = 6.51 Å, far from the 0.77 Å reported by LAMMPS.
Checked for periodic boundary effects: Issue persists even with PBC off.
Checked for simulation box size issues:
- Box size is large (55×55×55 ų).
- No atoms appear too close to the box boundaries.
Checked tabulated potential format:
- Used
pair_writeto generate a tabulated potential from LAMMPS itself. - Ensured same parameters for both
lj/cutandtable. - Issue only occurs when using the tabulated potential.
My Question
- Why does LAMMPS report an unphysical 0.77 Å separation only when using the tabulated potential?
- The same LJ potential runs fine when used directly but fails when read from a table.
- What could be causing this discrepancy?
I want to learn how to properly use tabulated potentials for a future project, so understanding this issue is critical.
Any help or suggestions would be greatly appreciated!
Here is the tabulated potential: # DATE: 2025-03-11 UNITS: real Created by pair_write# Pair potential lj/cut fo - Pastebin.com