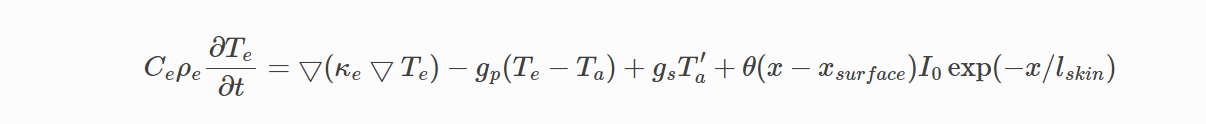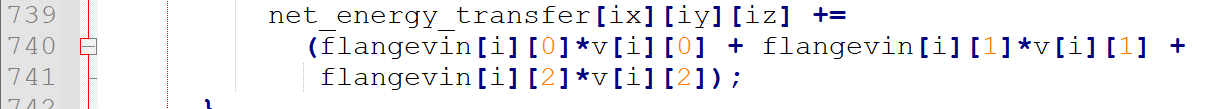Hello everyone.
Currently I am using lammps version 23/jun/2022 and
I am confused about the fix ttm/mod command, in the description document, the electron heat diffusion equation is solved in another grid that overlay the MD simulation box,
the grid and simulation box are futher divided into grid cells that models energy transfer between electrons and atoms.
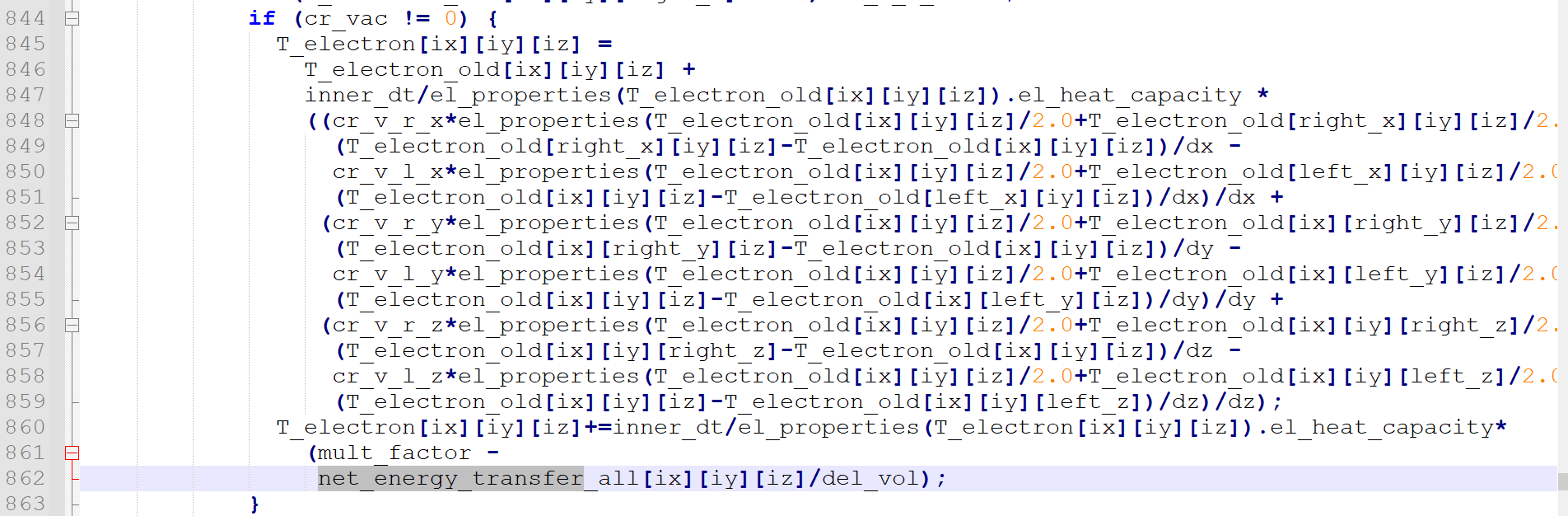
From source codd fix_ttm_mod.cpp, we can see that the temperature of each electron grid is computed from the 1. heat conduction from nearby grid cells.(line 844-859) 2. input energy(laer)(line861 mult_factor) 3. energy transferd from atoms(lin862, net energy transfer)
this is the most relevent information about net energy transfer all
Q1: I don’t understand how the net_energy_transfer_all is calculated, maybe it is calculated in another file?
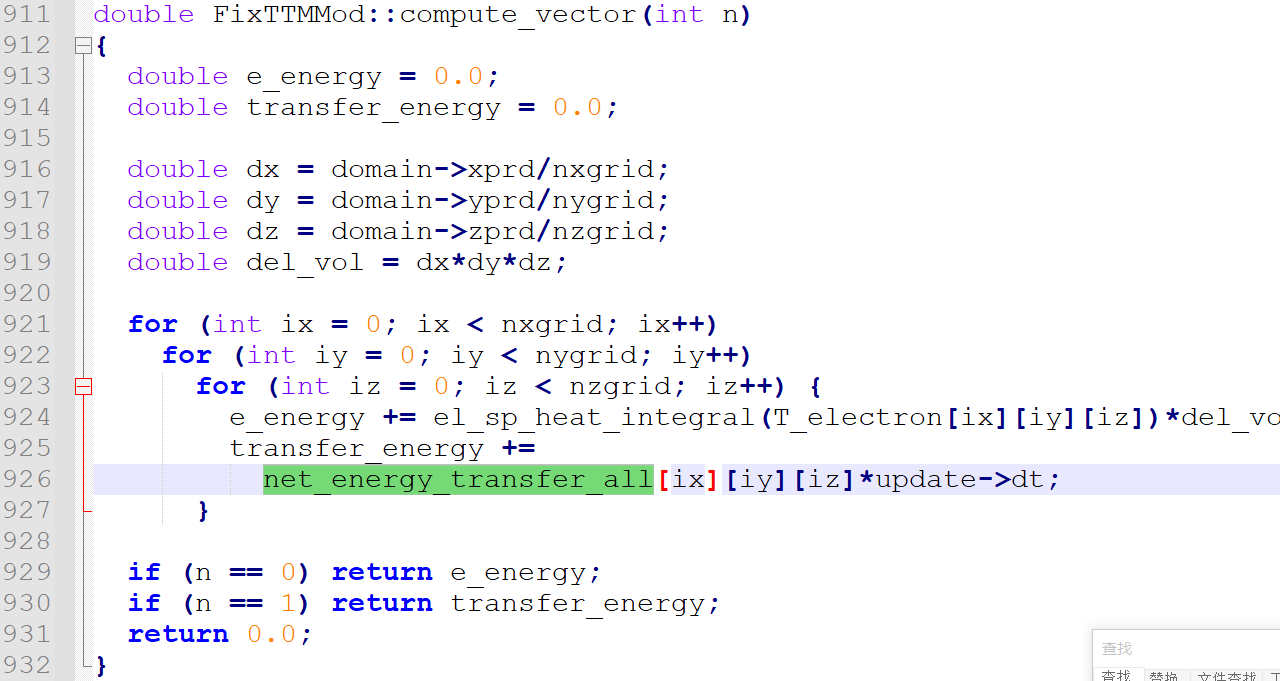
Q2:I don’t understand the function computer_vevtor(int n), can anyone explain it?
However, I do find some related codes to deeped my understanding about this command.
the energy transfer between electron and atoms are done through langevin force so I find the langevin force calculation term:
then using calculated per-atom force to determine energy.
Q3: the document illustrate that the energy transfer is down through through temperature difference between electrons and atoms in that grid(ignore electron stopping), but I can’s find the calculation of atoms temperature in that grid. Am I miss something?
Q4: how can I modify the code to get the atoms temperature at each grid cell in that timestep(not MD step)? @Steve@akohlmey
Can anyone help me @ authors, I am not sure if I used proper format and @ correct user.
Thanks for all of yout patience and time.
There is no such thing as a per-atom temperature. Temperature is a bulk property and only well defined for large systems. We compute a temperature in computer simulations despite the small system size under the assumptions that a) the kinetic energy is equipartitioned across all degrees of freedom and b) that for a system in equilibrium the system average is equivalent to the time average. If you look at the most extreme example, the classical harmonic oscillator, you have a situation where the
“temperature” ranges between 0 and 2xT. Coupling atomistic data with a continuum model is tricky business (certainly beyond my area of expertise).
In general, I would suggest to first experiment with fix ttm, which is much simpler and thus easier to understand and then also study the published literature. This is not likely something where will get more info than just some general level information that you could have obtained easily yourself. If you want to discuss the science behind this, you have to find a suitable collaborator with expertise in the field.
thanks akohlmey!
I understand per-atom temperature is meaningless and per-atom velocity is important. What I meat is that: as the equation in the first picture illustrates, the magnitude of transport energy between two system is determined by the temperatue difference and g_p,where is the per-grid temperature T_a? From your reply and the source code, do you mean calculating grid temperature of atom is tricky so that you used per-atom velocity multipy force(as force is a vector) to represent energy transfer? Am I correct?