I’m trying to simulate a 2D MOF structure with this published interatomic potential using the given minimal input and get the following warning:
WARNING: Bond/angle/dihedral extent > half of periodic box length
The initial structure is DFT optimized for cell dimensions and atomic positions. The topology file for LAMMPS is correct to the best of my knowledge.
The indications that something unusual is going on are:
Very high potential energy at the beginning
Distorted structure before and after minimization (i.e. bonds across periodic boundaries are either elongated or shortened)
Input:
units metal
special_bonds amber
atom_style full
boundary p p p
dielectric 1.0
kspace_style pppm 1.e-4
pair_style lj/charmm/coul/long 14.0 16.0
bond_style hybrid harmonic morse
angle_style harmonic
dihedral_style charmm
improper_style cvff
pair_modify mix geometric # default
read_data ${path}/DATA
include ${path}/potential.coeff
timestep ${dt}
thermo 100
thermo_style custom step time temp press etotal pe ke evdwl ecoul ebond eangle edihed eimp
minimize 1e-10 1e-10 10000 100000000
I’ve already done my research to my best capacity within this forum and came across somewhat similar posts (1, 2, 3, 4, 5) but unfortunately couldn’t apply the suggestions to my scenario.
I appreciate the insight of any expert so that I can begin to understand this issue and take relevant steps to correct it. I’m more than happy to provide further information if necessary.
How small is your system?
How did you create the data file and specifically assign bonds, angles, and dihedrals?
Can you provide a copy of the data file? …and a minimal input file?
There are 1223 atoms in the orthorhombic (rectangular considering only 2D periodicity) unit cell.
How did you create the data file and specifically assign bonds, angles, and dihedrals?
I used VMD topotool utility for creating the data file and assigning bonds, angles, and dihedrals. Custom atom names were specified in the PDB file manually before obtaining topology with topotool (I did my best not to make a mistake)
Can you provide a copy of the data file? …and a minimal input file?
TopoTools will not create bonds across periodic boundaries unless you add them explicitly with manual VMD/Tcl scripting. Angles and dihedrals are derived from bonds, so it should be possible to auto-create them after the bonds are properly assigned.
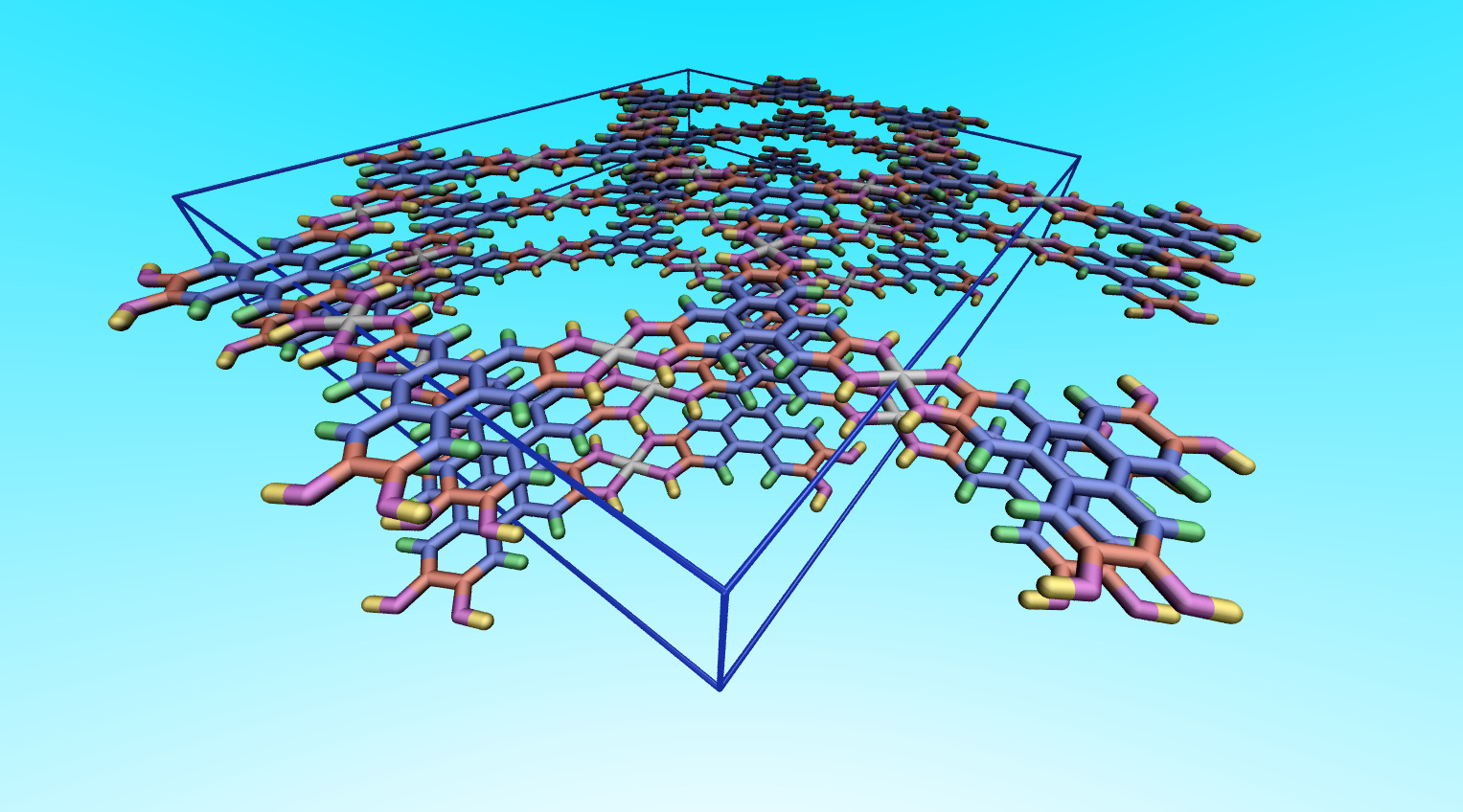
Yet, you didn’t even check the result properly. The box dimensions are completely bogus. You have overlapping atoms due to atoms outside the box and due periodic boundaries with atoms at the top and bottom of the box. See attached visualization of your data file from VMD:
Yet, you didn’t even check the result properly. The box dimensions are completely bogus.
I see. Seems like I have overlooked a couple of most important things. I thought giving the exact unit cell as in a periodic DFT calculation (i. e. VASP) would be correct.
Thank you very much for your time and patience checking my result!
I believe I now have a better understanding of what went wrong and what needs to be done. I’m just checking to ensure I’m on the right track. I understand that you are not obliged to clarify anything for me, but I would greatly appreciate any insights or comments you might have to offer.
Yet, you didn’t even check the result properly. The box dimensions are completely bogus. You have overlapping atoms due to atoms outside the box and due periodic boundaries with atoms at the top and bottom of the box.
So I corrected the structure to avoid overlapping atoms due to the reasons you mentioned. The structure now looks like this.
Additionally, to clarify regarding your second comment:
TopoTools will not create bonds across periodic boundaries unless you add them explicitly with manual VMD/Tcl scripting. Angles and dihedrals are derived from bonds, so it should be possible to auto-create them after the bonds are properly assigned.
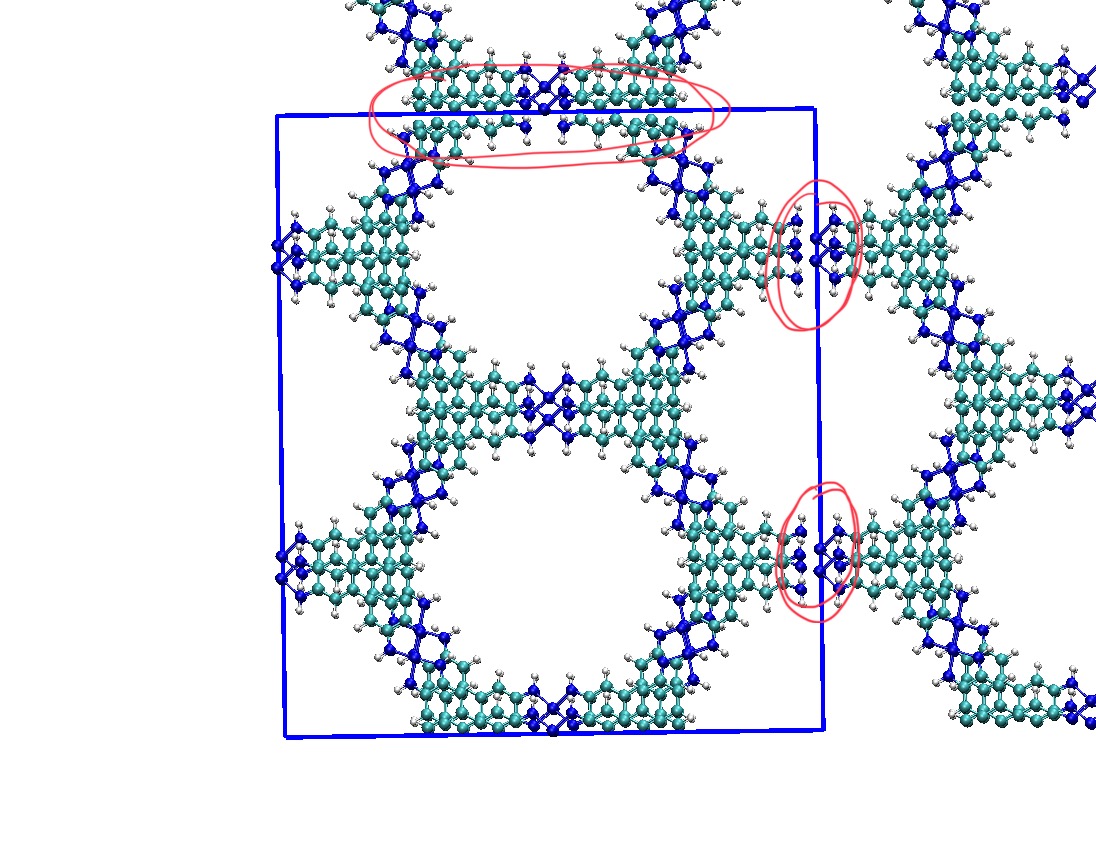
My understanding is that I need to use the ‘addbonds’ commands within VMD/Tcl scripting to add the missing bonds across periodic boundaries. Is that correct? In the VMD visualization, these bonds appear as within the structure itself rather than between periodic images. Is it safe to assume that LAMMPS will correctly interpret these bonds as connecting across periodic boundaries? In the attached image, I have highlighted in red the regions where I need to add bonds across periodic boundaries.
You are trying to turn this forum into serving as your tutor. That is not its purpose.
If you want to be certain about things like what you are asking about, just set up a tiny test input with only a few atoms and try out what you want to do and then check if it behaves as expected.
That is just part of how science works and a necessary skill to acquire. Soon you will need to be trying to do something where there is nobody around that can confirm whether what you are doing is suitable or not. So it is better to start making this a habit right at the beginning where the problems are small and rather easy to figure out.