I have compared the force-field parameters produced (OCCO_10k_in.data) to the OPLS paper as well as the above papers and the dihedrals, coefficients etc look correct.
I am initially attempting to equilibrate the system at 10k and then will run NVT to get the correct density and ramp T. However when I run the simulation the hydrogen on one OH collapses in to the other and the whole system explodes, which implies something is not correct
I have tried to optimize the starting positions of the atoms within OCCO_10k_STABLE.data and have got the system stable with one atom, however when I scale to two atoms the same thing happens, the system starts stable but eventually one hydrogen falls into the oxygen (see the log file).
Ideally i am hoping to run 1000 or so molecules, has anyone got any ideas or advice? I think perhaps the atom charges are the problems, but not sure how to modify / optimize these in a systematic way?
I guess I could also use a different force-field, but seems strange as OPLS has been used historically and so should work?
I forgot to add I have also tried running using ethanol as well as 1,3-Propanediol following the same procedure, generating the force field from ligpargen and both seemed to run fine, which seemed strange. Seems like specifically ethylene glycol is causing the problems. Thanks
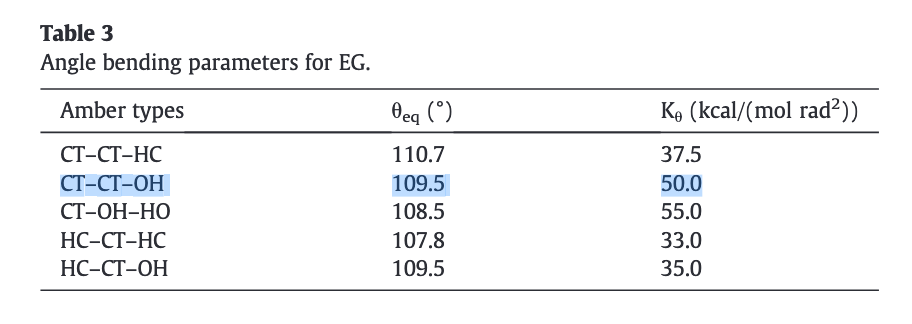
By looking at the dynamics of a single molecule, it seems that either the O-C-C angle or the H-O-C-C dihedral terms are missing or have the wrong parameters.
This is another case of “The stuff you have to figure out that articles don’t explicitly tell you about.”[1]
There are issues to have with LigParGen but as far as I am concerned, most of my problems were located between my keyboard and my chair when using it. Since it does not provide forcefield settings for LAMMPS, you have to figure them out yourself which is error prone.
Please have a look at this discussion. This should help you understand what is missing in your input file. I’ve no problem running a condensed phase with your molecule data file and the simulation doesn’t crash with proper settings of the forcefield. Let me advise you to double check your previous simulations with other molecules that supposedly “run fine”.
As a personal note I also tend to fix H covalent bonds with fix shake with timestep of 1 fs and higher.
As a side note, I am very skeptical with articles stating “We used forcefield XXX” without providing parameters of forcefield XXX in SI but a single ref in text because, sometimes, they actually didn’t use parameters of forcefield XXX or changed some stuff and only tell you if asked. ↩︎
Thank you so much to everyone who has replied and taken the time to try and help! Huge thanks to @Germain , you have solved the issue, the papers even mention coulombic scaling of 0.5, but not going to lie I did not know what that meant (and now do).
Are your referring to the section “8.6.4 Moltemplate Tutorial”?
Indeed, those settings are not explicitly mentioned in the tutorial. The reason is that the tutorial focuses on how to use Moltemplate to create a complete input deck for a LAMMPS simulation, including the DATA file.
Specifically, the OPLS-AA settings are inherited from the file oplsaa.lt but not reported in the manual. However, you will find them in the .init file.
@hothello I am referring to section 8.4. Force fields howto actually. Maybe extending the discussion of the AMBER FF since OPLS-AA was based on it at least initially.
It is a good suggestion. Actually, the settings for OPLS-AA are buried in the documentation of the pair_style hybrid/scaled command, Section 4.131
Note the global settings are effectively lj/coul 0.0 0.0 0.5 as required for OPLS/AA:
But I agree that it would be handy to have it also in S8.4, as the OPLS-AA force field is indeed supported by the command dihedral_style.
@akohlmey: I have added a brief section at the end of S8.4. Do I need also to add the new reference to the file Bibliography.rst? Howto_bioFF.rst.gz (4.3 KB)
Not exactly the topic of the thread, but I can second that statement:
I have got very weird potential parameters (including charges, which sometimes do not even sum to zero for supposingly neutral molecules) by using LigParGen. At least thats what it looks like when I compare it with the official OPLS-AA publication. It is always difficult to tell because you never know if version number 7000 of these general force fields has been released and thus things have changed, but I didnt find it very trustworthy.
If it ever helps someone in the future, I have made a python code that reads the necessary directives of the .itp files from GROMACS (which I assume is a trustworthy source for OPLS-AA potential parameters… if not, all is lost), gets the potential parameters for the atom types in hand and prints it in LAMMPS format for the sessions Pair Coeffs, Bond Coeffs, and so on of the data file (taking into account unit conversion and differences in potential forms from GROMACS to LAMMPS). If anyone ever finds it useful, I am giving the link: GitHub - cecimarques/gff_lammps: Python code to get force field parameters from GROMACS .itp files. Disclaimer: I am not advertising it - you dont even need to cite or anything - just sharing as I found it particularly useful since I need to run classical simulations using OPLS-AA for like 500 different organic liquids hehehehehe