Dear LAMMPS community,
I have a system with long range Coulomb interactions (as well as buckingham/mdf pairwise terms). I am trying to understand the effect of the two kspace parameters coul/cut and accuracy, such that I can decide on reasonable parameter settings for energy evaluations (ultimately I will be building a Monte-Carlo type simulation using the LAMMPS Python API, hence why I have included bonds and angles that are not used).
In order to try and choose these parameters I have tried evaluating the energy with different accuracies and varying the coul-cut (from very short-range, through to higher cut-offs). I’m not sure I expected the energies to diverge at higher cut-offs, as shown below:
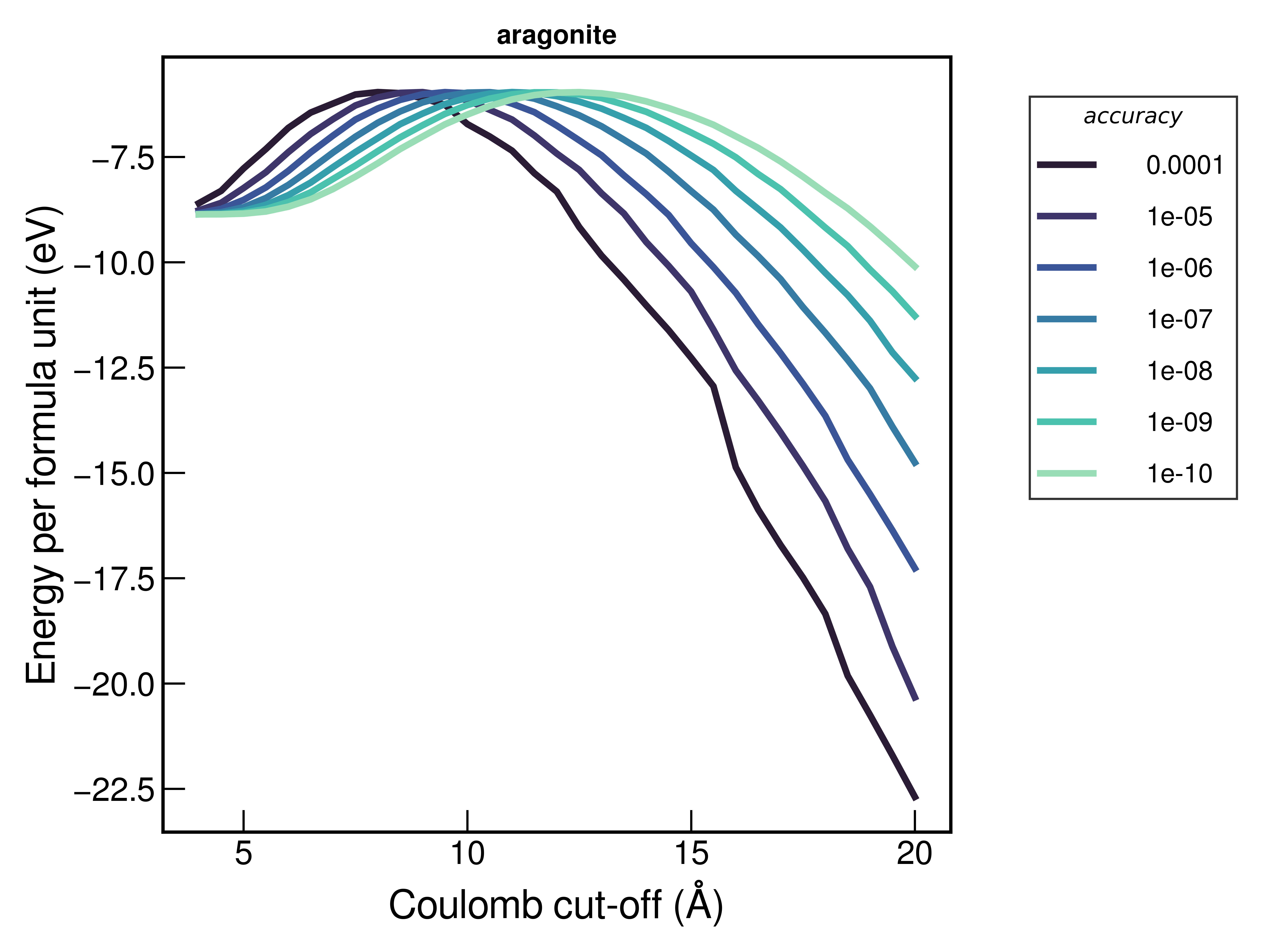
I also note that the absolute energies change depending on whether or not I include the bonds/angles information in the input file; does this occur because the default behaviour is to discount intramolecular contributions to e.g. Coulomb energy?
My data file and input script as given below. Any advise or suggestions would be appreciated. Thank you!
Thomas
“”"
custom lammps file
20 atoms
12 bonds
12 angles
0 dihedrals
0 impropers
3 atom types
1 bond types
1 angle types
0.0 5.7407000000000004 xlo xhi
0.0 4.9611000000000001 ylo yhi
0.0 7.9672000000000001 zlo zhi
Atoms
1 1 2 2.0 4.250758722000001 1.240275 0.6765746240000001
2 2 2 2.0 4.360291278 3.7208250000000005 4.660174624
3 3 2 2.0 1.3804087220000003 1.2402750000000002 3.307025376
4 4 2 2.0 1.4899412780000005 3.7208250000000005 7.290625376000001
5 5 1 1.123282 0.4889928260000004 1.2402750000000005 6.071882791999999
6 5 3 -1.041094 0.5486386990000004 1.2402750000000005 7.347670528
7 5 3 -1.041094 0.5009334820000004 0.13161798300000027 5.42287468
8 5 3 -1.041094 0.5009334820000003 2.3489320170000005 5.42287468
9 6 1 1.123282 2.3813571739999997 3.720825 2.088282791999999
10 6 3 -1.041094 2.3694165180000004 2.612167983 1.4392746799999998
11 6 3 -1.041094 2.3217113009999997 3.720825 3.3640705279999996
12 6 3 -1.041094 2.3694165179999995 4.829482017 1.4392746799999998
13 7 1 1.123282 3.3593428260000007 1.2402750000000005 5.878917208000002
14 7 3 -1.041094 3.371283482000001 2.3489320170000005 6.527925320000001
15 7 3 -1.041094 3.371283482000001 0.13161798300000033 6.527925320000001
16 7 3 -1.041094 3.4189886990000016 1.2402750000000002 4.603129472
17 8 1 1.123282 5.251707174 3.720825 1.8953172080000007
18 8 3 -1.041094 5.192061301 3.720825 0.6195294720000003
19 8 3 -1.041094 5.239766518000001 2.612167983 2.54432532
20 8 3 -1.041094 5.239766518000001 4.829482017 2.54432532
Bonds
1 1 5 6
2 1 5 7
3 1 5 8
4 1 9 10
5 1 9 11
6 1 9 12
7 1 13 14
8 1 13 15
9 1 13 16
10 1 17 18
11 1 17 19
12 1 17 20
Angles
1 1 6 5 7
2 1 6 5 8
3 1 7 5 8
4 1 10 9 11
5 1 10 9 12
6 1 11 9 12
7 1 14 13 15
8 1 14 13 16
9 1 15 13 16
10 1 18 17 19
11 1 18 17 20
12 1 19 17 20
“”"
“”"
# ---------- Initialize LAMMPS ---------------------
units metal
dimension 3
boundary p p p
atom_style full
# ---------- Create atoms ---------------------
read_data {fileName}
mass 1 12.011 # C
mass 2 40.078 # Ca
mass 3 15.9994 # O
# ---------- Define non-bonded interactions ---------------------
pair_style hybrid coul/long {coulCut} buck/mdf 6.0 10.0
#
pair_modify table 0
pair_coeff * * coul/long
kspace_style pppm {coulTol}
#
pair_coeff 2 3 buck/mdf 3161.6335 0.271511 0.0000000 #Ca-O
pair_coeff 1 2 buck/mdf 120000000.0 0.120000 0.0000000 #Ca-C
pair_coeff 3 3 buck/mdf 63840.199 0.198913 27.899008 #O-O
# ---------- Define Energy Calculation ---------------------
neigh_modify one 10000 page 100000
compute PE all pe
run 0
“”"