Hi LAMMPS community,
I have recently been working on a problem that requires the Langrangian binning of particles in a compressing molecular solid (making part of a thermalized sample a static piston and imposing a negative velocity on the rest of the sample to make the two collide). The way I have been attempting to bin the particles (and have the same bins correspond to the same particles) has been as follows:
variable samplebot equal bound(zmin,all)
variable top equal ${samplebot}+30
variable sampletop equal bound(zmax,all)
region piston block EDGE EDGE EDGE EDGE EDGE ${top}
region piston include molecule
region rest subtract piston
# I am writing this from memory so the regions might not be right, but this essentially is separating the #sample into a piston region 30 ang tall in the z axis, and the rest region which is the rest of the #sample.
compute rest chunk/atom bind/1d z ${top} 6 nchunk once ids once compress yes units box
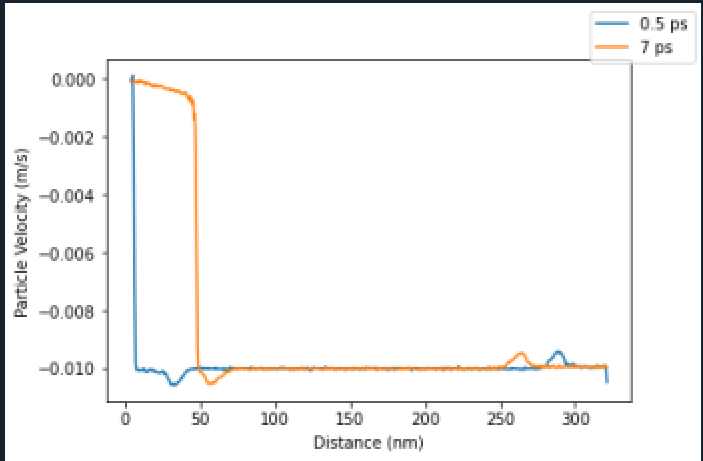
This seems like it should work fine (though the chunks won’t correspond to real distances once the ‘rest’ portion of the sample starting moving), since each atom only has its chunk ID assigned once. Thus, I would expect that regardless of where the atoms end up over time, they should still be part of the same chunk. However, when running these simulations there is a disturbance in all of the variables that I am computing using chunk averaging propagating from the free end (i.e. there is a increase in particle velocity near the free end at propagates at the imposed sample velocity, and an decrease near the piston surface). I have been going crazy with freezing variables, redefining bins, attempting to impose a dynamic bin size based on the amount of deformation the sample has endured, but nothing gets rid of these propagating disturbances in velocity, pressure, mass density, or strain near the free end of the sample. Further, these variables return to the expected value once the disturbance has passed.
The disturbance near the piston surface makes sense since there is probably some slop with how the binning origin is defined which causes the average to always include some atoms with zero velocity (thus raising the average), but I am totally at a loss with what is happening at the free end.
It seems to me that the intended functionality of the ‘nchunk once ids once’ phrase is exactly to define bins that follows the particles which it was initially defined on, and this seems to be the case when looking at the chunk/ave output files.
Has anyone done anything similar to this and been successful?