Here are my documents:
dimension 3
units metal
atom_style atomic
boundary p p p
timestep 0.001
read_data 5.data
replicate 10 10 10
pair_style none
compute 1 all xrd 1.541838 Cd Te 2Theta 20 90 c 0.01 0.01 0.01 LP 1 echo manual
fix 1 all ave/histo/weight 1 1 1 10 100 250 c_1[1] c_1[2] mode vector file 211Theta.xrd
run 0
In the case of a perfect CdTe single crystal, I tried a lot of output files in parametric cases, and none of the XRD plots were output in a single crystal style.
L=0
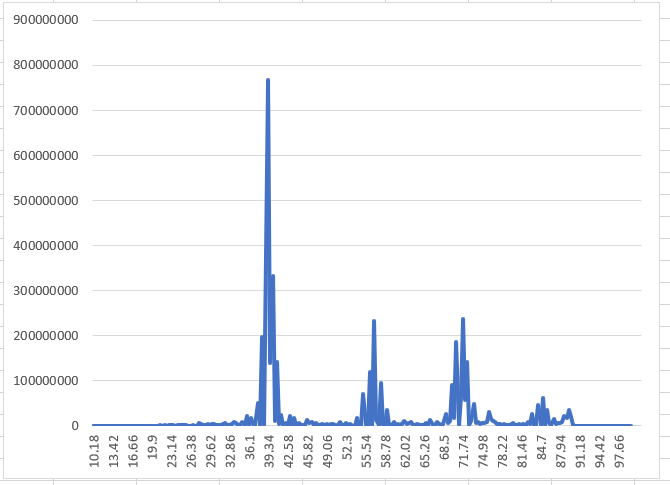
L=1
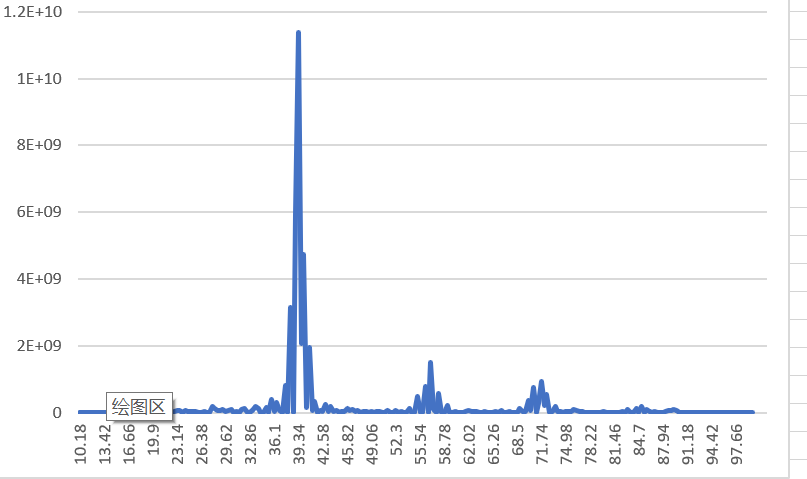
L=1 no manual c=1 1 1
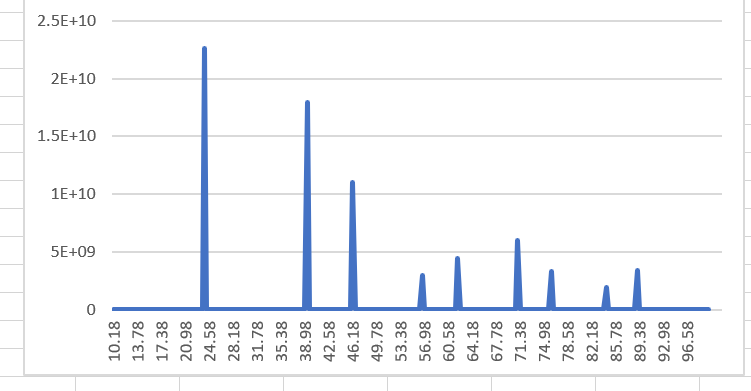
I also tried the file given by Lammps’ official website, and he is also a perfect single crystal, and the output XRD is still not a single peak.
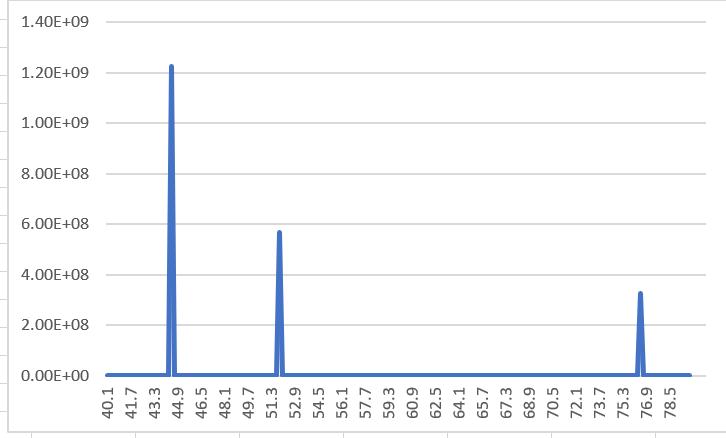
I would like to ask you how to get XRD images of single crystals.
Thanks!