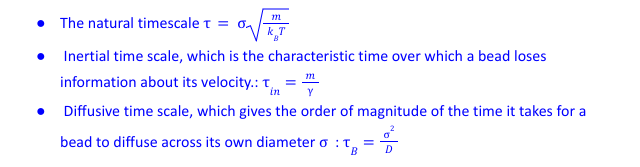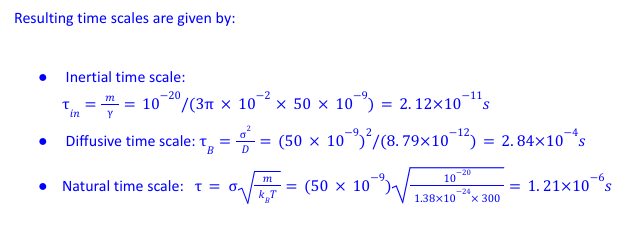I am trying to simulate a bead-spring polymer system with LJ 12-6 or WCA type interaction. There are three important timescale in the system:
Following previous published study on similar systems, I chose: m=\gamma=k_BT=1 which implied \tau=\tau_{in}=\tau_B.
In my system, m=10^{-20} kg, \nu=10^{-2} P, \sigma =50 nm~~ \text{and}~~ T=300K.
Clearly this shows the three time-scales are orders of magnitude different and \tau_{in}<<\tau<<\tau_B. Now For numerical stability one must choose the time step \Delta t smaller than all of these times i.e. \Delta t<\tau_{in}<<\tau<<\tau_B. However, I wish to study phenomena which will occur on times of the order diffusive timescales \tau_B. To avoid unfeasible long run times I keep \Delta t =0.005\tau_B \sim 10^{-6}s which ensures many steps occur within a typical diffusive time.
Now my questions are:
Does this ensure an overdamped conditions akin to low Reynolds numbers?
Can one trust that inertia is not palying any role in the dynamics occuring in such implementation?
Does LAMMPS use any additional method to make system efficient compared to raw velocity-Verlet? Running with \Delta t<\tau_{in} is practically impossible here.
If one is only interested in only long time behaviour (No dynamics) such as the final state after running 10^7 steps, will there be any backdraw due to the choice of m=\gamma=k_BT=1 and \Delta t =0.005\tau_B?
If I read this right, you are mapping your LJ \sigma to a length of 50 nm and you are mapping your LJ mass to 10^{-20} kg, which is 1E7 atomic mass units. Those seem like very large values to map onto a single LJ bead. Can you clarify how you derived those values?
I guess I’m still confused. Usually when we use sigma, tau and kT, we are talking about the Lennard-Jones parameters of the Coarse grained potential. But when you calculate your diffusion timescale, you refer to sigma as if its the overall size of your whole macromolecule. Are you coarse graining all the way to the point that there is 1 LJ particle per macromolecule?
The coarse-graining scale is 5000 basepair of DNA. generally a chromosome is of the order of 100 Mbp. So in these simulation, chromosome is modeled as bead-spring polymer where each bead is 5kbp.
This is a standard method in coarse grained polymer physics designed to work in mesoscopic scale. Here, atomistic level details are not being considered.
I think your issue relies mostly with how one goes back and forth with the reduced units in molecular simulation. This is not specifically related to LAMMPS but how you put down and explain your problem. I suggest you read this discussion on the topic where we explained in detail how to use reduced units and compare them back to physical results. You might see why your question 3 and 4 do not makes much sense without additional details.
The results you’ll get and the underlying physics will also depends on how you set you simulation up and which models (that also includes thermostat, integration scheme etc.) you use.
It will be hard for the discussion to go any further without any actual simulation detail and a clearer view of what you are trying to achieve.
I am not sure what you mean by those chained equalities; as your own calculation shows, these timescales are vastly different.
Which means that the answers to your questions are fairly straightforward and standard: since the inertial timescale is much, much shorter than the thermal and diffusive timescales, your system does not “remember” any inertial motion on the timescale over which it actually shows any meaningful thermal or diffusive movement; this does correspond to the “overdamped regime”, which in practice means that there will be negligible difference between Boltzmann dynamics and Langevin dynamics.
But the answers are not as important as whether you understand the answers and can do correct science with them.
Thanks all for the clarifications. These comments along with my own background reading have clarified many things which I was simply kept assuming for many days based on previous published works.
I want to take this chance to point out a relevant portion from lammps documentation:
Apply a Langevin thermostat as described in (Schneider) to a group of atoms which models an interaction with a background implicit solvent. Used with fix nve, this command performs Brownian dynamics (BD)…
I feel the part “performs Brownian dynamics (BD)” is little confusing. Fix langevin, along with fix NVE performs Langevin dynamics.
Now the Langevin dynamics can have different regime like underdamped or overdamped. Only overdamped langevin dynamics corresponds to Brownian dynamics, where inertia can be completely neglected and we drop the intertial term. I am doubtful if the underlying equation of motion contains the inertial term, whether it can be called Brownian dynamics directly.
Recent inclusion of “fix brownian” in the package enables to perform Brownian dynamics, because the equation of motion lacks the inertial term completely.
If you feel this way, please bring this to the attention of the LAMMPS developers by submitting a bug report issue on GitHub at: https://github.com/lammps/lammps/issues
I have not seen this mentioned in this discussion, so here are my $0.02 worth of thought:
You cannot run simulations on the larger time scales with a Lennard-Jones potential because of its repulsive core. A simple test to determine what happens would be to run an LJ system at the smallest time scale and then visualize the trajectory where you average the positions over N timesteps and vary N from small to large (in VMD this can be done automatically). You should see that when averaging over a large enough number of timesteps, particles will move “through” each other rather than around and this cannot be represented with an LJ potential. One method that would be suitable for this case is DPD (which is actually a whole family of methods).