Please, I couldn’t get thermo output in my log file. But I guessed there was no interaction within the atoms and neighbours. No potential error message. I change the time step from 0.001fs to 0.00001fs, yet no output. The simulation didn’t indicate finish time and no error message. I attached the log file for any assistance.
RSA_log.txt (7.7 KB)
in.Script (4.6 KB)
Your simulation seems to have stopped after a minimization step was completed, it looks OK to me.
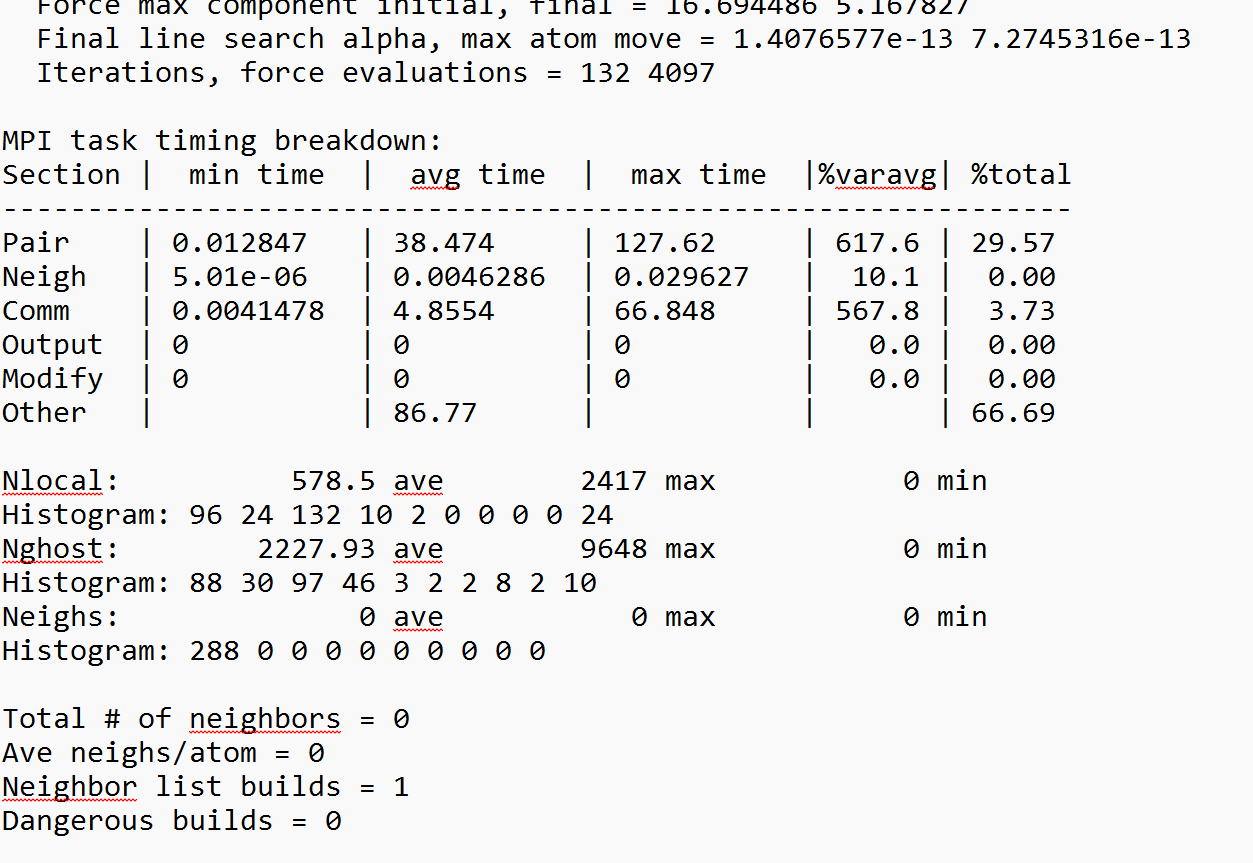
Per MPI rank memory allocation (min/avg/max) = 25.63 | 27.49 | 38.57 Mbytes
Step Temp E_pair E_mol TotEng Press Volume
0 0 -697941.49 0 -697941.49 79531.291 3355257.2
132 0 -749016.4 0 -749016.4 39812.276 3360670
Loop time of 130.1 on 288 procs for 132 steps with 166608 atoms
99.5% CPU use with 288 MPI tasks x 1 OpenMP threads
Minimization stats:
Stopping criterion = linesearch alpha is zero
In any case, you need to provide better details and read the forum guidelines if you want help here.
simon
@simongravelle. Thanks. Is the script that made it no to output the thermo? No total numbers of neighbour. In other simulation that worked to finished time. I have many number of neighbours unlike this that gave zero number of neighbour.
OK I see your input now…
I think that LAMMPS not writing anything during a “run” command can happen when the topology is really bad, may be due to some atoms overlapping or something. I would check the state of the system using VMD or Ovito if I were you.
Simon
That is because LAMMPS could not find a single representative neighbor list build for the whole system. You have a very complex system with hybrid pair styles.
Mind you, combining multiple EAM or MEAM pair styles with pair style hybrid like you do is a very bad idea. Your interactions of the different MEAM components will all have an incorrect embedding energy because of using a hybrid potential.
Another very bad idea is to use a potential with a cutoff shorter than (most of) the minimum potential energy distances like you do with the Morse potential. I would question the use of a repulsive-only potential regardless.