Hi
I am new to Lammps and have few very basic questions.
I am using Winmostar to run the simulations. To learn I started with simple molecules. I have done minimization, NVT and NPT equilibration. My queries are
-
Does the simulation end with this or does it have any production run? (I am relating it to different dynamics software like gromacs and Charmm)
-
I couldn’t find a complete tutorial for lammps, like a working example. Can anyone share a good set or tutorials or examples?
-
I didn’t get any dcd files during the simulation.
-
When i tried opening the lammps trajectory i get the structure as it appear in the attached file
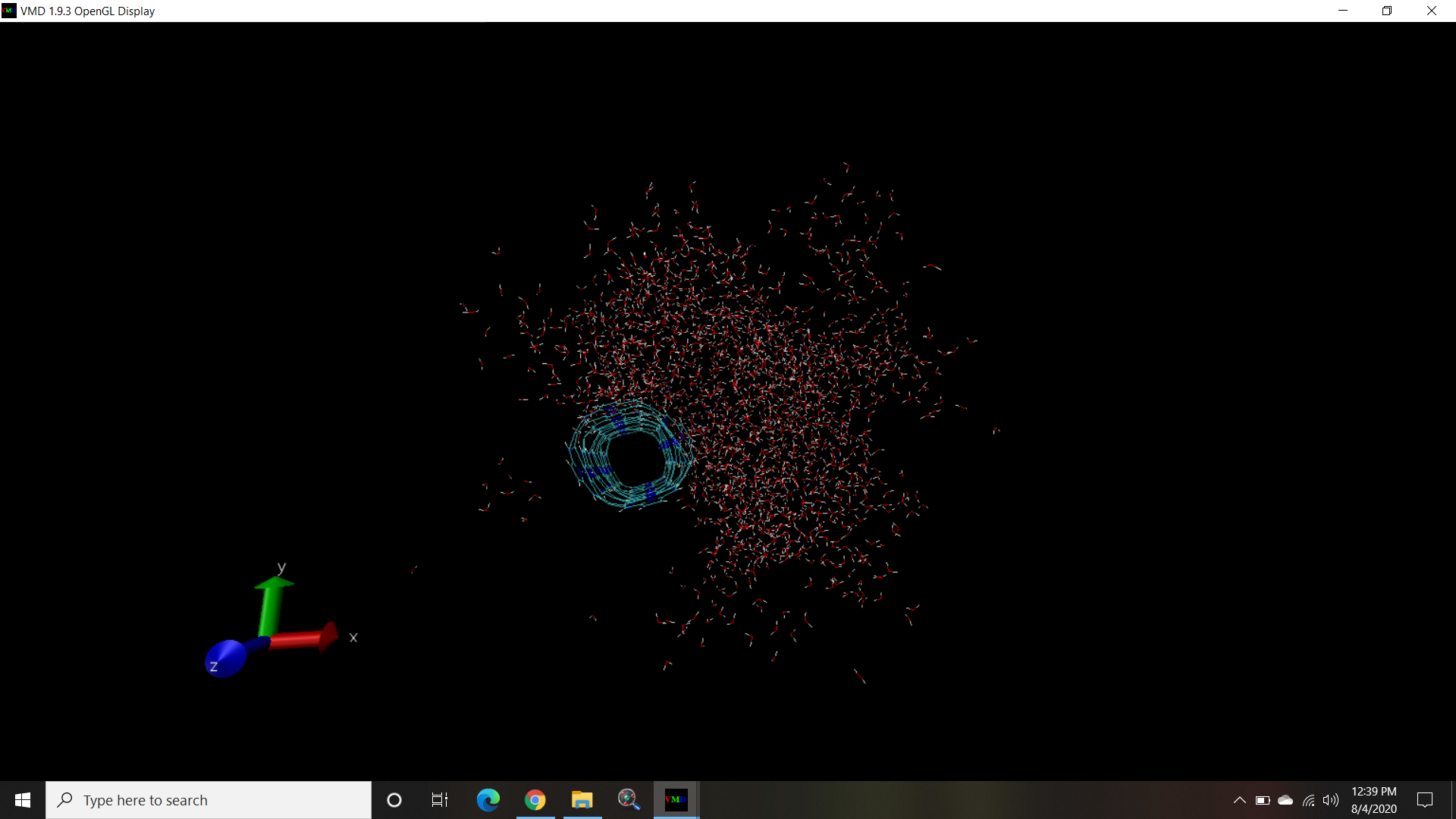
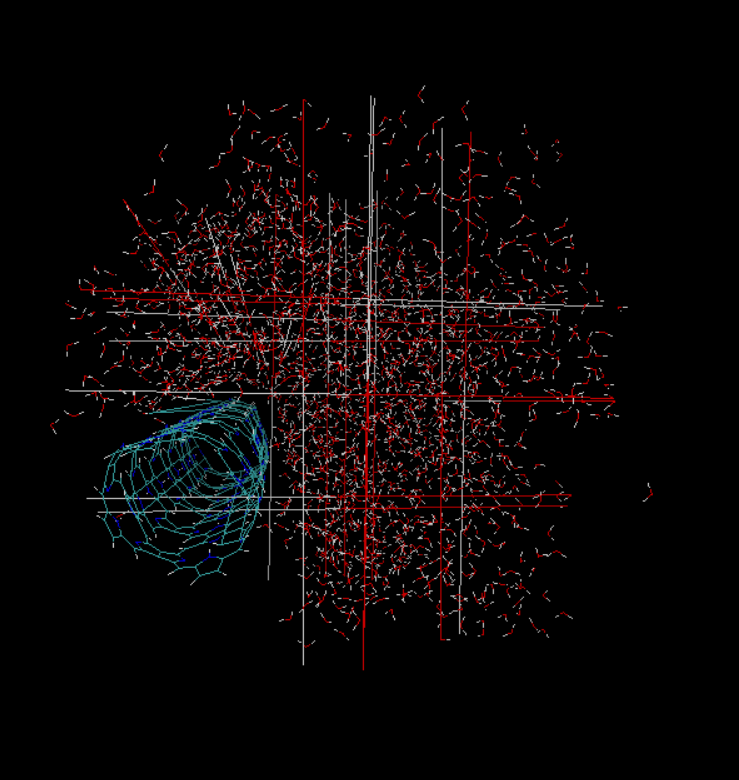
- When I tried opening the xtc file using vmd as we do for gromacs trajectory, I get these long bonds, which might be related to pbc. How to rectify it.
Spare me for such long mail.
Hi
I am new to Lammps and have few very basic questions.
I am using Winmostar to run the simulations. To learn I started with simple molecules. I have done minimization, NVT and NPT equilibration. My queries are
- Does the simulation end with this or does it have any production run? (I am relating it to different dynamics software like gromacs and Charmm)
you have to refer to a guide on how to properly do molecular dynamics simulations. there are text books and tutorials online, but that is outside the scope of this mailing list. what LAMMPS does is documented in the LAMMPS manual. reading tutorials is no substitute for reading the manual.
- I couldn’t find a complete tutorial for lammps, like a working example. Can anyone share a good set or tutorials or examples?
the LAMMPS manual has a detailed discussion on what happens when you run LAMMPS, it comes with examples. there also are tutorials that were given at various LAMMPS workshops and meettings. the material from the last LAMMPS workshop is available at: https://tutorial.lammps.org/
- I didn’t get any dcd files during the simulation.
to get dcd files you have to use the corresponding dump style in the input. details are in the manual.
- When i tried opening the lammps trajectory i get the structure as it appear in the attached file
there is no question here.
- When I tried opening the xtc file using vmd as we do for gromacs trajectory, I get these long bonds, which might be related to pbc. How to rectify it.
this is a VMD/visualization issue. apparently you have stored “wrapped” coordinates so if atoms belonging to a bond move across periodic box boundaries, you get this kind of image.
axel.

