It is not the app, it is the installer that needs to be programmed accordingly.
…and it requires Windows Vista or later, but that is pretty much a given for desktops/laptops these days.
It is not the app, it is the installer that needs to be programmed accordingly.
…and it requires Windows Vista or later, but that is pretty much a given for desktops/laptops these days.
I just uploaded new packages with version v1.3 of the LAMMPS GUI. This is dependent on features only in the LAMMPS development branch, so there will be no backport to the stable branch and v1.2 will be bundled with the first update.
New features and improvements of the new version:
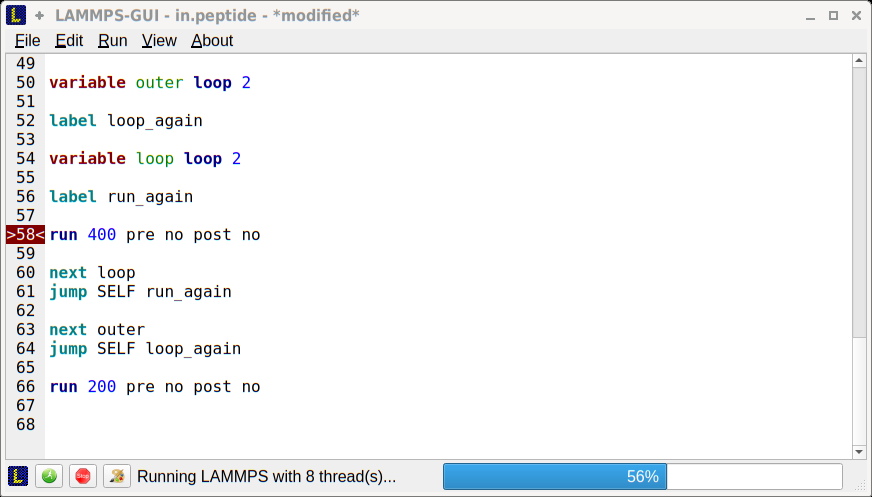
lammps_commands_string()jump SELF <label>) with lammps_commands_string()lammps_commands_string() one can send the filename and LAMMPS will read the file from disk (in case there are issues with loops and multi-line strings, reading from file should always work).info variables command. This command has been enhanced to also display the current value of loop and uloop style variables.So, a bit late, but here goes the results of my “testing” of the specific “functionalities” of the viewer for the v1.2 (Windows) that we were discussing some days ago:
zoom in/zoom out: seems okay
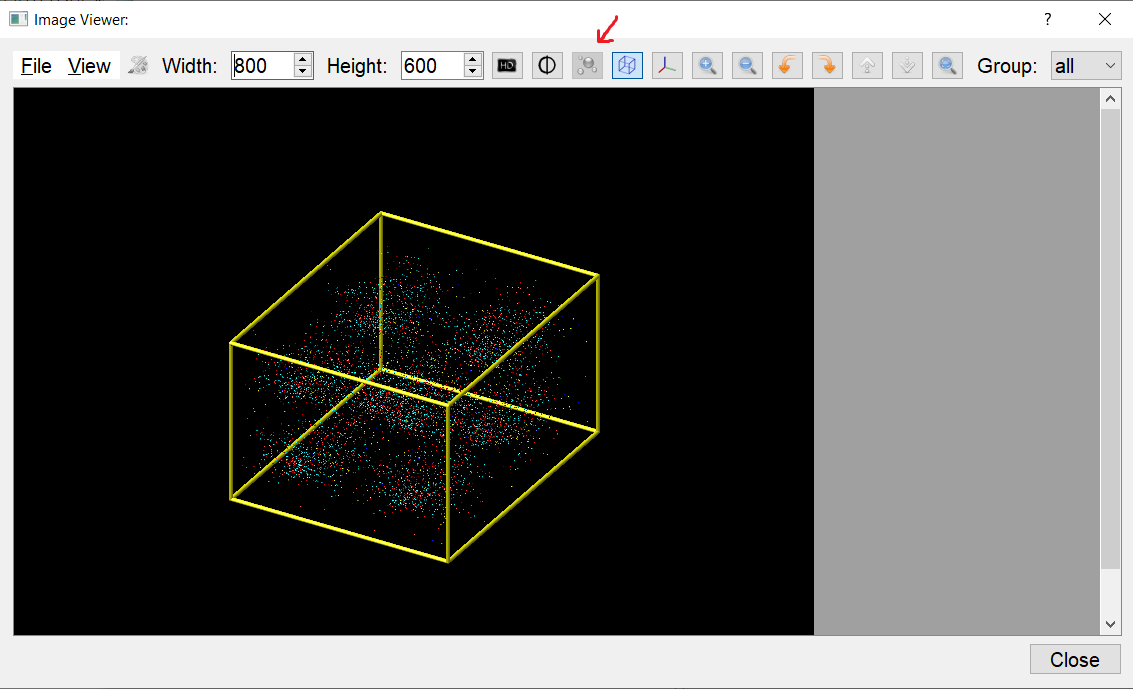
rotatation of the box: seems okay
display the edges of the simulation box: seems okay
display the axis: seem okay
display only specific group of atoms: seem okay
width/height : seem okay
SSAO rendering : seems okay
Anti-aliasing: seems okay (or at least it is doing something… I myself have a difficulty assessing this tool (but it is my problem, not the tool))
Pallete coloring while LAMMPS is busy: seems good also.
For the zoom, if there was some intention of making it work by means of us scrolling forward and backward the “middle-button” of the mouse (when it exists), it isnt working. Similarly, if there was an intention of being able to rotate the image by means of a click-hold-drag on the image (besides on the buttons of the image viewer), it is not working either.
I ran two different simulations: one with 5500 atoms and one with 19000. In my testing, for a simulation box of 5500 atoms turning on the HD is quite fast and so are the other functionalities in general. None of the functionalities seem to take “a lot of time” (defined here as “several seconds”, as stated in the LAMMPS GUI manual).
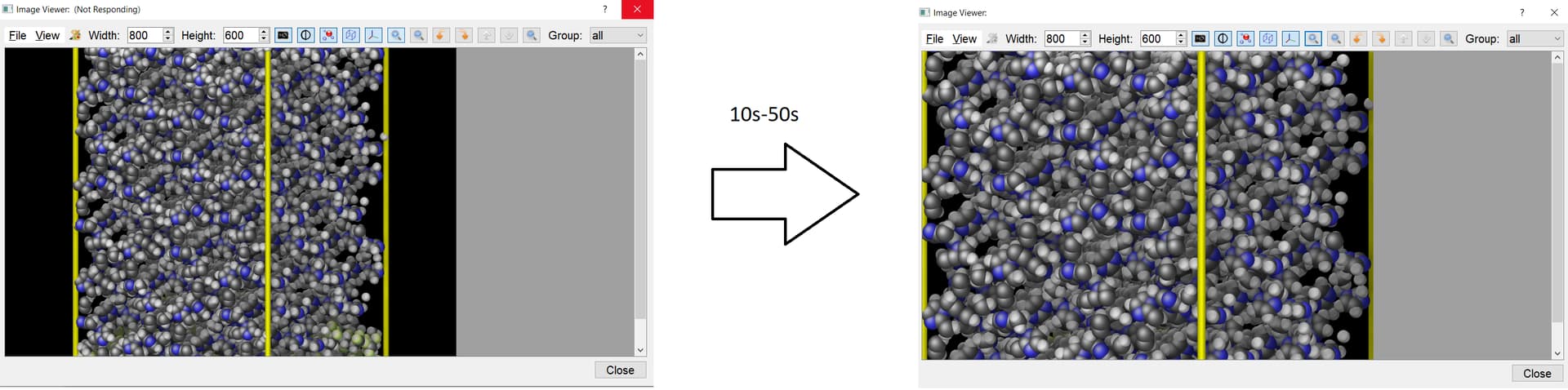
For a simulation box of 19000 atoms, turning on the HD takes a couple of seconds and the zooming in/zooming out can take a couple of seconds too. I find it quite tolerable though. However, for what is worth it, for the zoom-in/out, if the person clicks desperatly on the zoom button aiming to make it zoom in or out faster/(a lot), the program can go into “non-responding” mode for like 10-50s seconds and then it comes back to life with the job done. (e.g. shown below). Note that this is only for the 19000 atoms simulation box: for the 5500 atoms one it doesnt happen. But in any case this is reported in the manual and also, if the person waits a bit, the program comes back to life with the zoom-in/out done successfully.
I am not familiar with the “van der Waals style representation” (although I’ve seen it exist in VMD) and its possible “requirements”. For some reason, I am able to enable it for my simulation with 19000 atoms but for the simulation box with 5500 atoms the option is not possible. The button seems to be uncolored to me (as if I cannot toggle/untoggle it). I am showing a picture below. I couldnt figure out why it is possible to use it in one case and not the other. For what is worth it, in both cases the style I used is the atomic and the script is equally basic.
Finally, when I close the buffer in a case where I wrote things on it on my own (i mean, without having summoned a file which would be now modified), there is no message asking me if I want to save the “modifications” (into a file) before I close it. I dont know if it was indeed intentional (up to you to judge). In the manual it says it will only ask if you want to “save the modifications” in case you have modified a script that was originally saved in a file. So I suppose it’s good.
Dear @ceciliaalvares,
Thanks for taking the time to experiment and give feedback. Much appreciated.
There are some images with details posted in the manual
https://docs.lammps.org/latest/dump_image.html#image-quality-settings
Basically, this renders the image at 4x the size and then scales it down by computing the color of one pixel from average of a square of 4. This should result in much smoother edges. You see the effect everywhere these days when you read text in any graphical application.
There is no such intention.
It depends a lot on whether you have multi-threading enabled and the number of threads configured in the preferences. By default it should do, but if you have tried multiple versions of the app, there may be some leftover settings from a previous (buggy?) version and you should see what happens. HD mode (i.e. SSAO) is quite time consuming, especially in combination with anti-aliasing, since it scales worse than linear with the number of pixel. Anti-aliasing is at about 3x the cost; both together can be up to 20x. But I found a way to multi-thread the SSAO calculation, and that can cut down the time it takes significantly.
It is important to note that this is not supposed to be the ultimate visualization tool. But rather a quick and dirty way to get some image (through LAMMPS’ dump image command) and to make choosing the options for dump image more convenient. For proper visualization, there are options to delegate the data to either OVITO or VMD (through writing a data file and then loading it).
The requirements are: it has to be possible to identify elements so that covalent radii can be assigned and that requires that a) units are either “metal” or “real” and b) all atom masses can be matched with the masses from the embedded periodic table of elements.
That may be dependent on the platform and how you close it. In some cases it is possible to bypass the checks in the program. There should be a dialog to ask to save if there is a modified label in the main window title.
Ah, okay, then it’s all good (the “problematic” simulation was a coarse grained one, being the beads’ masses the sum of the masses of the atoms encapsulated)
I’ve uploaded new packages with version v1.4 of the LAMMPS GUI. This uses more changes in the core LAMMPS code from the development version and thus is based on a development snapshot.
New features and improvements of the new version:
Unless somebody has an idea for the must-have killer feature, this will likely be the go-to version for a while.
Update: In other words, anybody that thinks this is a useful program should download and try it on as many OSs and variants as possible and try to crash it and make it misbehave. I have already discovered a couple of cases where the syntax highlighting was missing some commands or syntax elements, so at least one more round of fixes is pending. But also suggestions for corrections and updating the “Quick Help text” or the “Howto manual” to me more useful to beginners would be highly welcome. It is very difficult for me to put myself into the mindset of a typical beginner, so what I consider important may not be that crucial. Keep in mind that I am “in the business of figuring out software”, so many things are obvious to me that are not to everybody. thanks again for any help and support. We will probably use the GUI for the next edition of the tutorial at a LAMMPS workshop, since it now provides all the functionality required for performing the exercises.
Feedback, bug reports, constructive criticism, suggestions for features are welcome.
I can test all the new features for v1.4 for windows and also see again if I can push it to crash or something this weekend. I will also try to read the manual with more “critical eyes” to give my possible suggestions for possible-improvements to make it easier for noobs (maybe I am suitable as a noob ^^)
That is great. On the weekend there will already be a version 1.5 with line reformatting and completion. The first is (already have it working for commands, i.e. the first “word” in each line). Pull request is already in the works: Add reformatting of line and command completion to LAMMPS GUI by akohlmey · Pull Request #3912 · lammps/lammps · GitHub
I am currently rushing to get everything done as far as possible, because in the next two weeks, I will be lecturing at an HPC workshop on Colombia and during that time I won’t have access to the (virtual) machines that I am using for building the binary packages.
Update: a not yet complete version 1.5 has dropped an hour ago. This has (auto-)completion only for commands. The rest needs a little more work. Since I have done auto-completion already with the lammps-shell code using readline, I don’t have to figure out the basic logic, just adapt it from using readline to QCompletion in the Qt library.
The v1.5 version I posted this morning has something like that for commands, support for available styles if forthcoming. With the default settings, after you have typed the first 3 characters of a command, a popup should appear with possible completions. Moving the cursor down to select on of them with enter will complete the command. The auto-completion can be switched off in the editor preferences and turned into a manual completion on pressing Shift-TAB. This also features an auto-reformat when typing Enter/Return after a line is completed or typing TAB any time. The various width settings can be changed in the preferences. BTW: this updates the list of commands (and styles) automatically to those available in the given executable at run time.
This is appears to be a specialty of macOS (not the only one I have come across). For now there is a workaround to switch into the individual user’s home directory. That choice could be augmented, if desired in the preferences dialog. I’ll have to think about it. In most OS cases, the user home is going to be the default, so for now, all OS variants should behave the same.
Comments from my side so far:
I had no problem using the dump image command within my different attempts to use the command and the slide show viewer works just fine (although my test is with the .exe, not with the .zip). So everything should be fine for the .exe at least
Now with this functionality of the slide show, you unlocked the possibility of “visualizing a trajectory” which I suppose was the one possible missing point for using LAMMPS GUi as a tool for begginers to learn about MD and etc ^^
I did not find any weird problem in any of the tests I did, although I admit they were not as many as before. I didnt find anything that would seem to me as some miscoloring of the text either.
PS: I dont know if it was something you did or if my computer is somehow magically performing better, but I am no longer able to make LAMMPS GUI go into “non responding” mode if I click on the zoom/unzoom very fast. In fact it responds quite well to my clicking in terms of speed!
For the manual, I’ve just finished reading it again and no idea of improvement popped up in my head… it seems all quite understandable… maybe if I let it marinate for some days something can come up…
Dear Axel,
Again, your efforts are much appreciated! I installed and ran a few examples on the v1.5 you’ve posted for macOS. Here is my system software and hardware information:
Software:
System Software Overview:
System Version: macOS 12.6.8 (21G725)
Kernel-Version: Darwin 21.6.0
Boot Volume: Macintosh HD
Boot Mode: Normal
Computer Name: Tapashree's MacBook Air
User Name: Tapashree Pradhan (tapashreepradhan)
Secure Virtual Memory: Enabled
System Integrity Protection: Enabled
Time since boot: 17 days 15:52
Hardware:
Hardware Overview:
Model Name: MacBook Air
Model Identifier: MacBookAir7,2
Processor Name: Dual-Core Intel Core i5
Processor Speed: 1.8 GHz
Number of Processors: 1
Total Number of Cores: 2
L2 Cache (per Core): 256 KB
L3 Cache: 3 MB
Hyper-Threading Technology: Enabled
Memory: 8 GB
System Firmware Version: 481.0.0.0.0
OS Loader Version: 540.120.3~22
SMC Version (system): 2.27f2
As before, all the steps from the README executed perfectly.
The auto-completion feature works perfectly!
Also, I have found out that if I change the directory in my Terminal and run the lammps-gui command, then the working directory does change easily (very sorry ![]() for that suggestion, it is straightforward to do from the Terminal itself).
for that suggestion, it is straightforward to do from the Terminal itself).
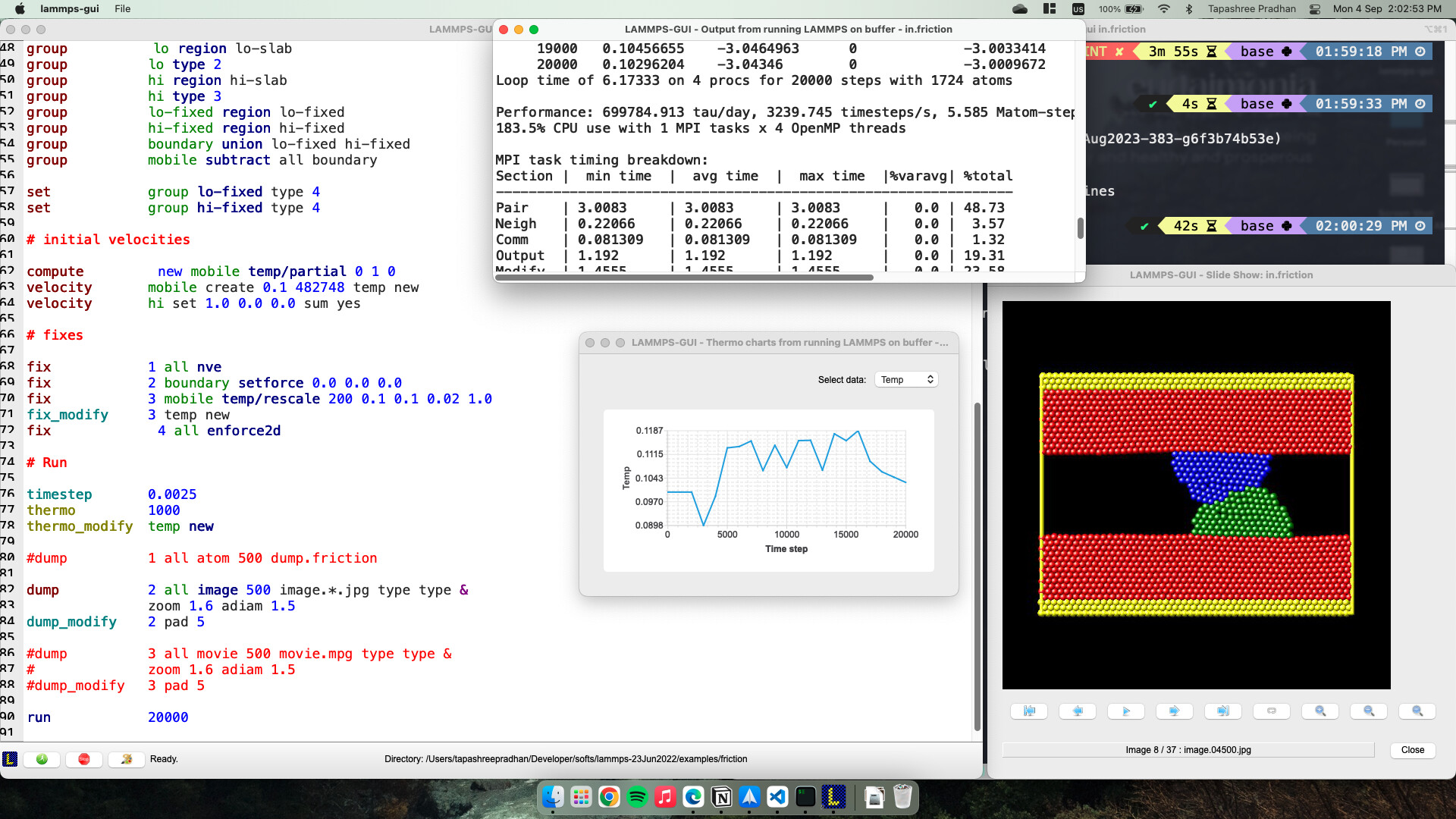
I also ran a few scripts from the examples folder of the LAMMPS 23Jun22 version build that I had already configured in my system. Below, you can see the screenshot of the in.friction script.
The start/stop buttons are much appreciated! The slideshow window also worked perfectly, and the bottom panel reflects the directory change, too.
Perhaps the most unique feature of the GUI version you’ve built is when I run an input script, the graph and trajectory visualization window also open up simultaneously along with the log file, and I can see the progress without the hassle of opening up any other graphing/visualization software as the script continues to run. This will be pretty useful to me when I’m starting to write a new script and trying to study deformations and use fix commands I’ve not previously used and do not quite know what the thermodynamic/trajectory output should look like when the simulation is done correctly.
I do not have any more improvements/features to suggest, as I think the GUI has become quite functional and seamless. Thank you so much!
Warm regards,
Tapashree
Thanks for your kind words. Just keep playing around and let us know, either here or as a bug report issue on the LAMMPS GitHub site, if you can make the application crash or behave unexpectedly. Also, if the syntax highlighting is missing something or creating inconsistent colorization. In some cases, this cannot be avoided due to the (simplified) logic of how the colors are being determined, but as I was looking at more inputs, I occasionally found some corner cases, that had not been properly addresses, but could be corrected with some small tweaks to the existing highlighting logic.
Similarly, it should be possible to add highlighting to a few more cases, after I add a “syntax check” for an input. This would run the input, but skip over run commands. That would indicate syntax errors and would allow to know about which groups, fix and compute IDs, variables etc. exist and correspondingly suggest them in the auto completion in cases where such a completion could be added logically.
Please let me remind you for that specific use case of the features available in the context menu. If you have typed the name of a command, you can look up its documentation on the LAMMPS website from the context menu. I found it quite useful personally, since for some commands I keep forgetting the exact syntax and the context menu helps to quickly find it. Normally I always have a tab in the web browser open with this page: 5. Commands — LAMMPS documentation and then open the corresponding sub-page and look up the documentation for a command from the (alphabetically sorted) table. Now this becomes just a single mouse click and selecting the desired entry from the context menu (or just hitting Ctrl-? (or Command-? on macOS).
New binary LAMMPS GUI packages (v1.5.4) with the latest bugfixes and improvements have been posted. These should have command completion working to about the same level as LAMMPS shell except for filenames. There are some principal differences between the completion mechanism used that currently prevent implementing this.
This should complete all intended features for a v1.x series of the app.
Planned features for v2.x are a complete rewrite to get rid of a few initial bad choices and have a better structured and more maintainable codebase. In the process, a port to support QT 6.x is planned.
The major new feature for v2.x would be a collection of “Wizard” dialogs which would enable setting up some standard calculation types by answering a few questions and providing some files. Again, I am looking for suggestions and (ideally) collaborators on this. This could also be an opportunity for machine learning enthusiasts to put “ML magic” into the app. While don’t believe the the currently available tools are useful to create whole input files, they seem quite suitable to provide suggested answer to some of the questions such a wizard system would ask and people may not be able to answer instantly and an ML based bot could gather some information from the web and its “memory” and provide some possible suggestions.
So, I am going to report a “bug” that I, myself, dont think is important because the person using lammps gui can realize on her/his own and manage super well without any problem whatsover. So I am reporting just in case it happens to be important for you.
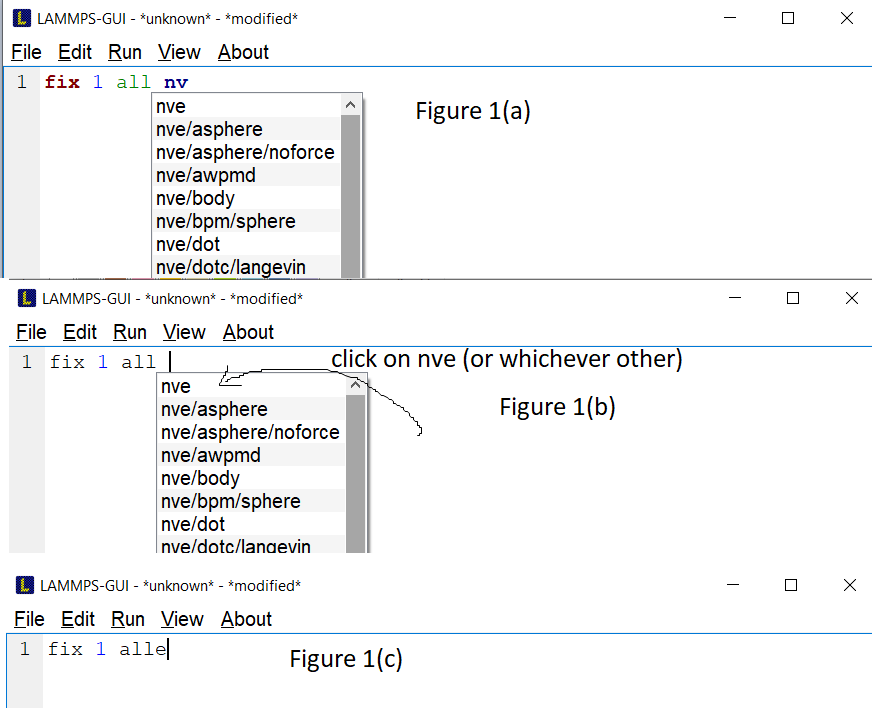
If you are typing the name of a given fix (or compute or etc), you can see the list of suggested completions, as shown below in figure 1(a). If I change my mind and erase the two letters I had written (the ‘nv’), the suggestions dont go away (figure 1b). And then, if I decide to click on one of these suggestions (say the fix ‘nve’, but could be any other) at that point, the completion adds the missing letter, which would be only the ‘e’ at the moment the screen of completions popped up, and deletes the space between the fix name and the group-ID (figure 1c).
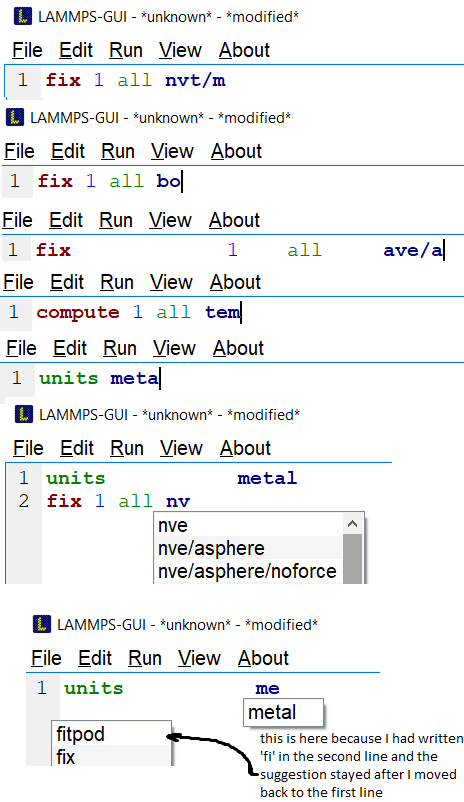
Then, if I realize that this is not what I wanted to do and erase the ‘e’ and go back to start typing again, the completions dont show anymore. Even if I erase literally all the characters of line 1 and start typing another command (like compute or units), I no longer see any suggestion for completion after typing two letters (I suppose you changed from three to two). However if I finish the writing by myself and press enter to go to the next line, the completions reset back to normal! Even if I go back to the first line and erase some letters now it offers me to complete again as expected! You can see all I wrote in this paragraph in the screenshot below.
PS: You can also note that as I erased everything in the second line to go back to the first, the suggestion I had once I had only ‘fi’ on line 2 remains there even though I am back to line 1. I suppose this is not expected either, but really it is 0% big deal…
PS: probably this was here since the previous version but I did not notice it… my bad ![]()
It is, in fact, a real bug. This is not the intended behavior. I copied the code that does the text insertion from some example and am now realizing that it is not behaving correctly.
I am changing how the completed text is inserted and this seems to give a much more consistent behavior now. It also allows to use a different mode of displaying possible completions. Rather than showing only those completions starting with the first two letters, it now shows the entire list with the most likely completion highlighted (according to the heuristics of the completer class, that knows nothing about LAMMPS). This seems more beginner friendly, since it makes it easier to change your mind. E.g. for units it will show all possible choices for units, even if you already typed “re” or “me” and there would be only one choice. I have been looking for a way to highlight the default completion, so one can just type enter to complete. With the “reduced” mode one has to use the mouse or the cursor keys to first highlight the choice (even if there is only one) and I found that a bit irritating.
So, many thanks for the report. You triggered two improvements to the code with one report. Well done. ![]()
New binary LAMMPS GUI packages (v1.5.6) with the latest bugfixes and improvements have been posted. Most of the changes were to increase portability and consistency. In addition, the following new features are included:
New binary LAMMPS GUI packages (v1.5.7) with the latest bugfixes and improvements have been posted. This latest set of changes is based on user feedback (many thanks to Keneth and Joaquin from the HPC School in Colombia!) and aimed to improve the experience and convenience for new users.
${gui_run} in file names and then have a new file for each run (so they can be later comparedNew binary LAMMPS GUI packages (v1.5.8) with the latest bugfixes and improvements have been posted. This mostly fixes incorrect behavior, programming errors, some crashes in some corner cases, and bugs versus the previous version. In particular:
New binary LAMMPS GUI packages (v1.5.9) with the latest bugfixes and improvements have been posted. The following significant changes are included:
New binary LAMMPS GUI packages (v1.5.10) with the latest bugfixes and improvements have been posted.
Outside of bugfixes, there are only two new features this time:
I won’t have access to the build servers for the next 6 weeks. It should be pretty difficult to crash LAMMPS GUI by now. Please post a reply here, if you manage it anyway and describe how you did it.