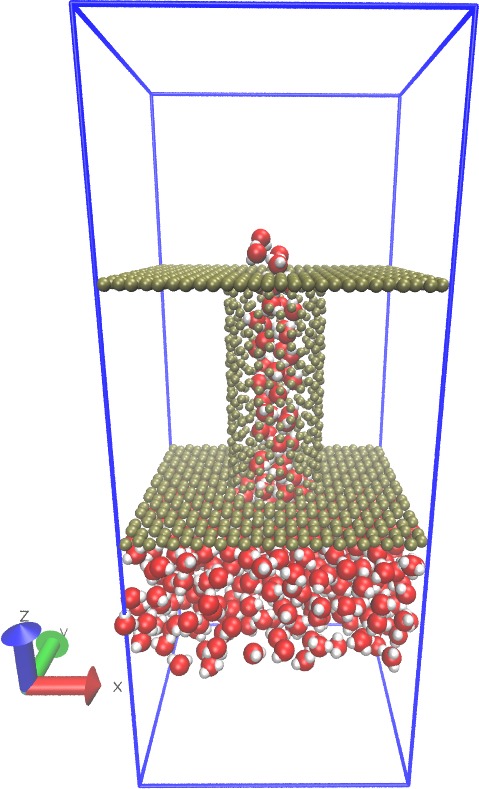
Hi,
I am using LAMMPS for MD simulation of carbon nanotube (CNT). I am new to this field and a beginner of LAMMPS as well. I have been trying to simulate water transport through CNT with the similar setup as in Ref. 1 & 2.
I am facing few problems with generating data script for LAMMPS of the initial system(with CNT, water and graphene sheet). I am using “VMD and Topotools (VMD plugin)” to generate the data script for a box of graphene sheet filled with water molecules whereas the box itself is connected with CNT. I have collected a script of SPC water molecules wrapped in a rectangular box and I can generate data script for CNT and graphene sheet separately using ‘Nanotube Builder plugin’. I can merge them together to create a single data script as well (according to the method described in here: https://sites.google.com/site/akohlmey/software/topotools/topotools-tutorial---part-2). However, the main concern is that I could not be able to make the geometrical system as described in those references especially how to connect the CNT and graphene sheet with a cylindrical hole in it (graphene sheet) as the same diameter of CNT or filling the CNT with water molecules (for another case). Is it possible to do using VMD and Topotools or TCL script (although still working on it)?
Please suggest me what would be the best (or usual) way to generate the data script for this kind of system using VMD (as I have been learning this). I have started using this for few months. I would really really appreciate if you could go easy on me. Any sort of help, suggestions/suggested readings/tools, opinions, are highly appreciated.
Thank you very much for your valuable time.
Ref. 1: http://prl.aps.org/abstract/PRL/v102/i18/e184502
Ref. 2 : http://jcp.aip.org/resource/1/jcpsa6/v137/i4/p044102_s1
Sincerely,
Ayub