I have been trying run a LAMMPS input and data file but have been getting the following error before job termination:
WARNING: System is not charge neutral, net charge = -9.1524 (../kspace.cpp:327)
I suspect there is something wrong with the input data file but I am not able to pinpoint the problem. From visual inspection, it seems fine as shown in OVITO below. I also revised the units, bond/angle types/definitions in the input file. I have attached the input file, data file, and error log to this message.
From the warning message, it should be straightforward: you have to check the assigned charges in the data file. Obviously, they don’t add up to zero as they should.
I was able to fix this charge issue but am getting a persistent low energy value after my energy minimization step which I think is not physical. After this, the calculation fails to make any progress.
Per MPI rank memory allocation (min/avg/max) = 17.55 | 17.76 | 18.16 Mbytes
Step Temp E_pair E_mol TotEng Press
0 0 2206.8984 137425.92 139632.82 7433.0915
28 0 -3.8817363e+15 129788.95 -3.8817363e+15 -2.5052522e+15
Loop time of 0.798277 on 48 procs for 28 steps with 900 atoms
I tried to revise my input script but was not able to find immediate errors. I have reattached the corrected input and data files (visually looks similar to before) as well as the complete output log from this run below.
Your input suggests, that your interactions for MoS2 are from a different source than your parameters for water. Has the MoS2 potential been parameterized and previously tested for interacting with water. I don’t see any explicit parameters between atom types 3+4 and atom types 1+2.
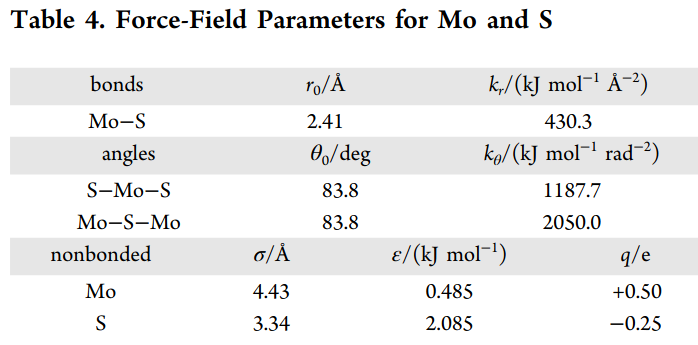
Thank you for the catch on the epsilon and sigma parameters @akohlmey! Yes, the MoS2 potential has been parameterized and tested for interacting with SPC/E water in the following paper and shown in the picture below:
This is how I get the bond_coeff and pair_coeff for MoS2 (making conversions from kJ to kcal) and using the bond_coeff and pair_coeff for SPC/E water model from LAMMPS SPC/E example. All of these are defined in the input script:
# Use this paper for force field parameterization (reaggregation section):
# https://pubs.acs.org/doi/10.1021/acs.jpcc.7b00484
# Bond interaction for MoS2 (per paper) and H2O (SPC/E)
# Format is as follows: btype (defined in .data) K(kcal mol^-1 Å^-2) r_0(Å) *converted to real units*
bond_style hybrid harmonic zero
bond_coeff 2 harmonic 102.8 2.41 # Mo-S bond
bond_coeff 1 zero 1.0 # H-O bond
# Define the angle interactions within MoS2 (per paper) and H2O (SPC/E) *converted to real units*
angle_style hybrid harmonic zero
angle_coeff 2 harmonic 489.96 83.8 # Mo-S-Mo angle
angle_coeff 3 harmonic 283.87 83.8 # S-Mo-S angle
angle_coeff 1 zero 109.47 # H-O-H angle
# Cutoff distance for both Lennard-Jones (L-J) and Coulombic interactions are set to 1.2 nm = 12 Å
pair_style lj/cut/coul/long 12.0
# Define nonbonded L-J parameters for Mo-Mo, S-S, and Mo-S interactions
# Format is as follows: atom_coeff atom_coeff ε(kcal/mol) σ(Å)
# Note that for the last pair_coeff we use Lorentz-Berthelot rules: https://en.wikipedia.org/wiki/Combining_rules#Lorentz-Berthelot_rules
# where σ_ij = (σ_ii+σ_jj) / 2
# and ε_ij = sqrt(ε_ii*ε_jj)
pair_coeff 3 3 0.116 4.43 # Mo-Mo
pair_coeff 4 4 0.498 3.34 # S-S
pair_coeff 3 4 0.240 3.885 # Mo-S
# Define nonbonded L-J parameters for H-H, O-O, and O-H interactions
# Using SPC/E water ff model, LAMMPS example here: https://docs.lammps.org/Howto_spc.html
pair_coeff 2 2 0.1553 3.166 # O-O
pair_coeff 2 1 0.0 1.0 # O-H
pair_coeff 1 1 0.0 1.0 # H-H
I am still getting the very low energy value after energy minimization. Other than correcting the epsilon and sigma parameters, everything is the same as the reply above. Would you be able to help me debug this issue?
I think what Axel means is that we aren’t convinced that this is a bug in LAMMPS vs bad input parameters. If you find a bug in LAMMPS then we are happy to debug if you provide a minimal working example to reproduce the issue, but we can’t do your research for you, which includes evaluating different force fields.