Oct’19 - Dear users, I have done a lammps simulation in Lammps.But when I put the lammpstrj file in OVITO , my kerogens are all out of the box. Could anyone help me ? Thanks a lot.
Hi @Yiquan_Jiang,
This looks more like a screenshot of the default VMD settings to me, not OVITO. These are not the same software. Are you sure about the one you are using?
If I remember correctly, unlike OVITO, VMD does not enforce wrapping the atoms at the periodic boundaries by default when loading dump files. Please see the documentation related to the commands you can use on their website, here for VMD and here for OVITO. These are basics of molecular simulation so you might want a colleague/advisor to teach you more about it face to face.
If you have more trouble in using VMD or OVITO, please post in the related mailing-list or forum section.
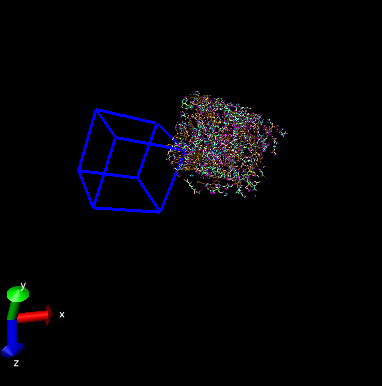
The picture was obviously created by VMD and not OVITO. The problem is a VMD issue. VMD was designed for use with NAMD and CHARMM and the DCD file format. That format does not store the origin of the simulation cell, only its dimensions. Unfortunately, this carried over to VMD so that it cannot know where the origin of a simulation cell is located.
When you use the pbc box command to draw the simulation cell, you have to provide the information about the origin explicitly. That information is lost when importing the trajectory file.
That said, you have periodic boundary conditions, so your the location of the box boundaries are pretty much irrelevant. You can verify from looking at the trajectory file itself (it is a text file) and see that the positions are well within the box.
yes,you’re right.It’ my fault.What I use is VMD ,not OVITO.Thank you for your reply!
I got it, thank you for your reply! ![]()
Dear akohlmey, there is a problem now. When I put my final data file in OVITO and I choose the UNWRAP TRAJECTORY, my kerogens are still out of the box. If this option is unchecked, a single atom and similar key breaks will appear inside the box due to period boundary. How can I avoid this and make my kerogens display completly or is there any problem with my script? I’m distressed about it and sincerely look forward to your reply. ![]()
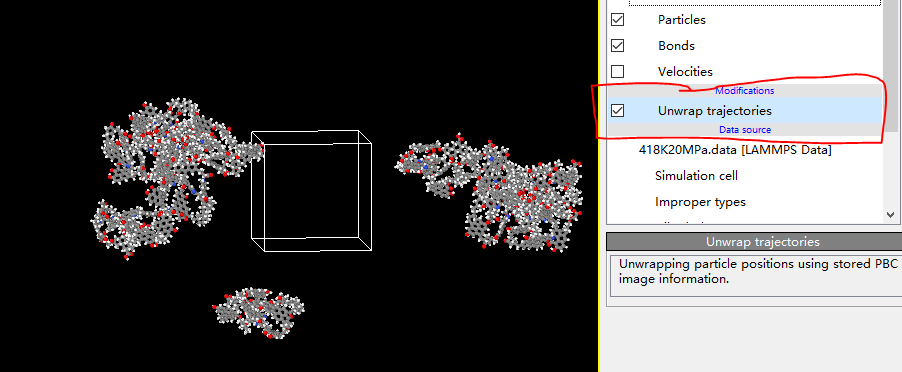
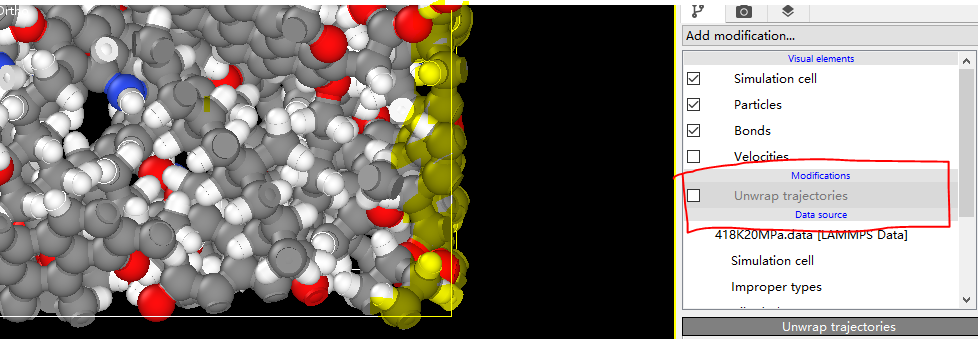
What you see is consistent with how the wrapping is executed in Ovito, and is completely correct.
If you want to wrap the molecules without breaking them, you can either write you own script that applies the minimum-image convention to a single atom per molecule (or to its centre-of-mass, if you want to be more sophisticated), or just load your trajectory in VMD and use the PBCtools:
pbc wrap -comp res -all
Of course, you need to have information about the molecular topology, i.e. at least a molecular index.
Thank you for your reply! There is still a question here. It means that I can use the matrix to build my model without any changes? Look forward to your reply.
What “matrix” and what “changes”? You are speaking in riddles here…
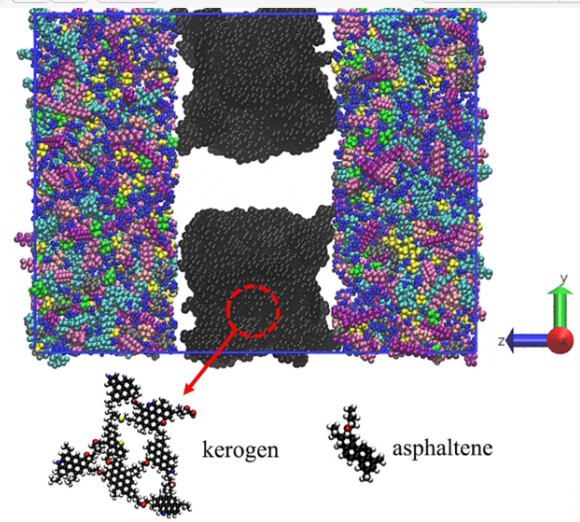
As the first picture shows, I also want to use the kerogen matrix to build my crack model and that’s what “matrix” means. The “change” means that I don’t know if I can use my matrix to build crack without using any methods to make my matrix display completely like the first picture shows. What I want to achieve is the situation in the picture.
As has been pointed out multiple times in this discussion. What you are worrying about is a visualization problem, not a system setup or simulation problem.
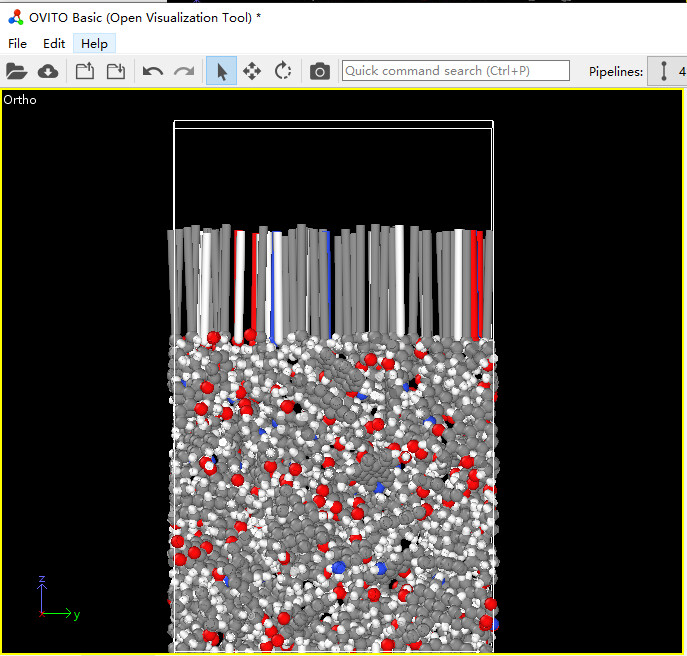
This is a completely different issue than what you described earlier.
Uh, my fault. At first I thought the problem was caused by software display issues.Because what I think is that if the matrix can be fully displayed, this problem will not occur. So could you give some suggestions for solving this problem?
If you want assistance with a new problem, you should create a new topic with a suitable subject line and provide sufficient information to understand what the issue is. Right now there is no explanation.
Since your original problem has been resolved, I will close this specific discussion topic now.