Dear lammps user,
First, my lammps version is 7 August 2019 .
I wrote the below input script which is assumed to generate a system of two dimensional sphere interacting through Lennard-Jones potential. I want to consider NVT ensemble. Therefore I fixed the reduced temperature to be 1.11 . However, LAMMPS outputs temperature which starts at 1.11 but then it changes quite dramatically during the simulation. Also the total energy of the system is not conserved since it does not have a constant value for the whole simulation.
I attached the plots of temperature vs. timestep and the total energy vs. timestep .
Could you please tell me what is the reason for NOT having constant temperature and constant total energy during the simulation ?
Much thanks in advance.
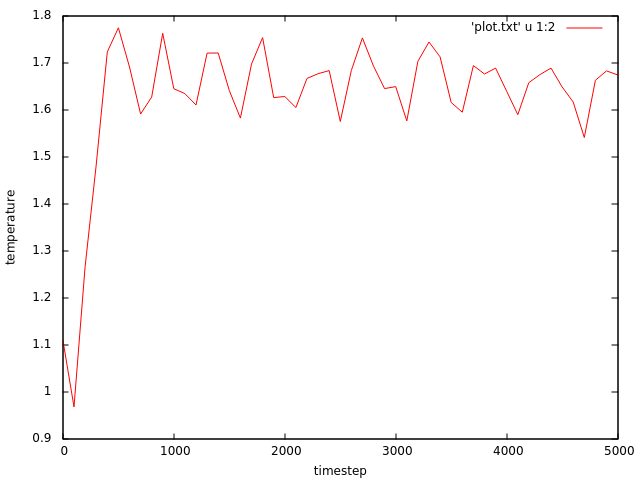
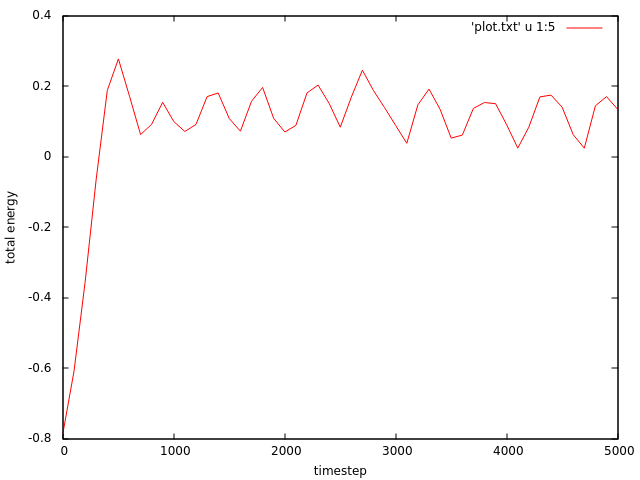
a) temperature on typical system sizes will never be “constant”. it will oscillate around an average value. the size of the fluctuations depends on the size, density, interaction model and parameters, and temperature of the system. this has been discussed without end on this mailing list and you can look up more detailed discussions in the mailing list archives and your favorite text book on statistical mechanics.
b) the temperature that is output as “thermo” output is by default only the temperature computed for point particles, not extended particles. to get the correct temperature for your system, you have to define a temp/sphere compute and monitor that, and/or use thermo_modify to replace the default compute temp instance with your new instance of compute temp/sphere.
axel.