My lammps version is lammps-23Jun2022, and I want to run MD simulations with ML hdnnp, and then use plumed to do the enhanced samplings.
I encountered a issue when I run the MD simulations:
terminate called after throwing an instance of ‘PLMD::Plumed::ExceptionError’
what():
+++ PLUMED error
+++ at Communicator.cpp:89, function void PLMD::Communicator::Set_comm(const PLMD::TypesafePtr&)
+++ message follows +++ you are trying to use an MPI function, but PLUMED has been compiled without MPI support
terminate called after throwing an instance of ‘PLMD::Plumed::ExceptionError’
I just use the make command to complie all things and have no errors generated for this complie process. I briefly describe the installation below:
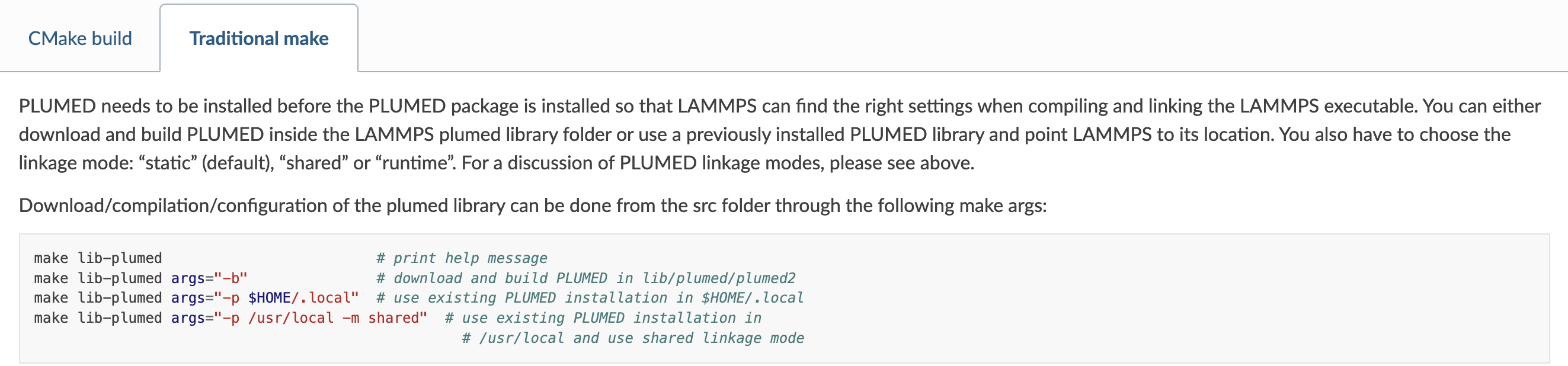
I first run: make lib-plumed;
then run: make lib-plumed args=“-b”
then run make yes-ml-plumed
then make mpi
I finally obtained executive lmp_mpi successfully and I perform MD simulation using the next command:
mpirun -np 20 lmp_mpi < xxxx.in
Unfortunately I encountered the error. It seems that this error tells me my plumed software cannot run multiparallel simulation, but I do not know what should I do to solve this error. Could you give me some advices? Thanks a lot.
You have to look very carefully at the output that you get during compilation.
The plumed configure script should find your MPI library automatically. It does for me:
Building plumed ...
configure: Optional modules are disabled by default
configure: Enabling all optional modules
checking for mpic++... mpic++
checking whether the C++ compiler works... yes
checking for C++ compiler default output file name... a.out
checking for suffix of executables...
checking whether we are cross compiling... no
checking for suffix of object files... o
checking whether we are using the GNU C++ compiler... yes
checking whether mpic++ accepts -g... yes
checking for gcc... gcc
checking whether we are using the GNU C compiler... yes
checking whether gcc accepts -g... yes
checking for gcc option to accept ISO C89... none needed
checking for gfortran... gfortran
checking whether we are using the GNU Fortran compiler... yes
checking whether gfortran accepts -g... yes
configure: Initial CXX: mpic++
configure: Initial CXXFLAGS: -O3
configure: Initial CPPFLAGS:
configure: Initial CFLAGS: -g -O2
configure: Initial LDFLAGS:
configure: Initial LIBS:
configure: Initial STATIC_LIBS:
configure: Initial LD:
configure: Initial LDSHARED: mpic++
configure: Initial SOEXT:
checking whether mpic++ accepts -fPIC... yes
checking whether gcc accepts -fPIC... yes
checking whether mpic++ accepts -Wall... yes
checking whether mpic++ accepts -pedantic... yes
checking whether mpic++ accepts -std=c++11... yes
checking whether mpic++ declares c++11 support... yes
checking whether C++ library supports C++11 exceptions... yes
checking whether mpic++ can generate dependency file with -MM -MF... yes
So you have to make certain that your environment module for MPI is always loaded and that you load the same module when you do “make lib-plumed args=-b” and when you do “make mpi”.
The alternative would be to try compiling with CMake. That should give you more feedback but be functionally effectively the same. It just compiles everything in one step.
This is a plumed library error and not a LAMMPS error. It seems to be compounded by using conda for your software management, something that I see a lot and thus recommend against it.
For properly using LAMMPS with PLUMED it is generally better to first install plumed separately and then point LAMMPS to that installation if this doesn’t happen automatically. Using the automated download and installation only links with the plumed library and does not provide access to the plumed command and other useful plumed features. It is mainly included in the LAMMPS distribution for testing that LAMMPS compiles with the PLUMED package.
This is the command that is used to compile the plumed library, but your machine setup is the reason it doesn’t compile.
Let me repeat, this is not a LAMMPS issue, but a plumed issue and thus not something I have an interest in debugging. Everything I wrote in my previous post still applies and is all that I have to comment about this.