Dear Dr. Shan,
Thanks so much for your encouraging advice!
1.Stopping at a step that is one step less than the multiples of fix reax/c/species’ Nfreq value is a typical error of fix reax/c/species. I already suggested you to try the Kokkos version.
I re-compiled lammps executable with Reax/c and kokkos packages. However, when I got the following error when I run the simulation with Kokkos package. How to solve this error?
ERROR: Cannot yet use minimize with Kokkos (…/minimize.cpp:57)
Last command: minimize 1.0e-4 1.0e-6 100000 1000000
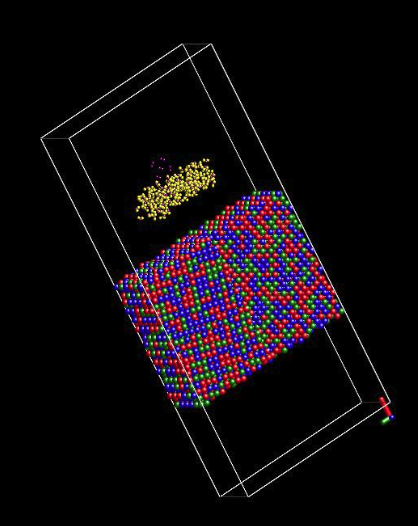
2.Your alloy substrate is a block of atoms isolated in vacuum. Instead you should have a slab or surface geometry. Please consult with local experts if you need further assistance in this regard.I re-built a bi-crystal Ni-Cr-Fe alloy as shown in the attached picture.
This time, by the following commends in red, I only expend the box in x-direction, while the y and z surfaces of alloy were set to match the boundary of box by the following commends. How do you like this slab alloy model?
lattice fcc 3.52
region box block 0 15 -10 10 0 6 side in
create_box 5 box
region left block INF INF -10 0 INF INF side in
lattice fcc 3.52 orient x 5 -9 0 orient y 9 5 0 orient z 0 0 1
create_atoms 3 region left
region right block INF INF 0 10 INF INF side in
lattice fcc 3.52 orient x 5 9 0 orient y -9 5 0 orient z 0 0 1
create_atoms 3 region right
read_data data1.watervmd add append offset 3 0 0 0 0 group medium shift 58 -15 0.5
read_data data4.oxygen add append offset 4 0 0 0 0 group medium shift 73 0 8.8
change_box all x scale 2 y scale 1 z scale 1
3. How did you specifically “switch off the H-Ni, H-Cr, H-Fe, O-Ni, O-Cr, O-Fe bonds in the force field file by setting the corresponding bonding parameters to 0 0 0”? Which parameters did you change? Changing force field file is a tricky business – are you sure you have changed the correct parameters?
I did the changes only in the following part of the force field file. For the other parts, I changed nothing!
44 ! Nr of bonds; Edis1;LPpen;n.u.;pbe1;pbo5;13corr;pbo6
pbe2;pbo3;pbo4;n.u.;pbo1;pbo2;ovcorr
bonds
before
after
H-Fe bond
2 4 78.2669 0.0000 0.0000 0.4668 0.0000 1.0000 6.0000 0.1766
0.5673 1.0000 0.0000 1.0000 -0.1543 5.4965 0.0000 0.0000
2 4 0.0000 0.0000 0.0000 0.4668 0.0000 1.0000 6.0000 0.1766
0.5673 1.0000 0.0000 1.0000 -0.1543 5.4965 0.0000 0.0000
O-Fe bond
3 4 67.5128 0.0000 0.0000 0.1301 -0.3000 0.0000 36.0000 0.0852
1.0000 -0.3500 15.0000 1.0000 -0.0629 7.1208 0.0000 0.0000
3 4 0.0000 0.0000 0.0000 0.1301 -0.3000 0.0000 36.0000 0.0852
1.0000 -0.3500 15.0000 1.0000 -0.0629 7.1208 0.0000 0.0000
O-Ni bond
3 6 74.5453 0.0000 0.0000 0.5189 -0.2000 1.0000 16.0000 0.0100
0.8623 -0.2500 15.0000 1.0000 -0.1416 5.2421 1.0000 0.0000
3 6 0.0000 0.0000 0.0000 0.5189 -0.2000 1.0000 16.0000 0.0100
0.8623 -0.2500 15.0000 1.0000 -0.1416 5.2421 1.0000 0.0000
O-Cr bond
3 9 114.0666 0.0000 0.0000 0.2305 -0.3000 1.0000 36.0000 0.6591
0.5793 -0.3500 15.0000 1.0000 -0.1989 4.8803 1.0000 0.0000
3 9 0.0000 0.0000 0.0000 0.2305 -0.3000 1.0000 36.0000 0.6591
0.5793 -0.3500 15.0000 1.0000 -0.1989 4.8803 1.0000 0.0000
Please have a check, thanks again!
Best regards,
Xiaolong