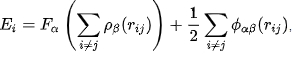Dear All,
I use LAMMPS version: lammps- 7Aug, 2019. I downloaded it from lammps website.
I was having difficulties replicating the values of potential energy of the system using the theoretical calculations and LAMMPS. I did it for three different systems. To do a sanity check, I tried to get the output of pair potential values for all the systems used, to my surprise these values were different from what was given in all the potential file.
I tried to calculate the potential energy for a 2 atom system which is described as:(For simplicity, I have shown only one of the input atom coordinates)
FeNiCr:
2 atoms
3 atom types
0.0000 5.0035 xlo xhi
0.0000 5.0035 ylo yhi
0.0000 2.3586 zlo zhi
Masses
1 55.845
2 58.6934
3 51.9961
Atoms
1 1 0.0000 0.0000 0.0000
2 1 2.87056 0.000 0.0000
The potential files were downloaded and replicated for research purposes from:
https://www.ctcms.nist.gov/potentials/
The LAMMPS input format looked like the following:
Find minimum energy fcc configuration
Mark Tschopp, 2010
---------- Initialize Simulation ---------------------
clear
units metal
dimension 3
boundary s s s
atom_style atomic
read_data atoms1.dat
---------- Create Atoms ------------------
---------- Define Interatomic Potential ---------------------
pair_style eam/alloy
pair_coeff * * FeNiCr.eam.alloy Fe Ni Cr
pair_write 1 1 5000 r 0.00112 5.6 table.txt RITEEEESH
neighbor 2.0 nsq
neigh_modify every 1 delay 10 check yes
---------- Define Settings ---------------------
compute eng all pe/atom
compute eatoms all reduce sum c_eng
---------- Run Minimization ---------------------
reset_timestep 0
thermo 10
thermo_style custom step pe lx ly lz press pxx pyy pzz c_eatoms
dump 1d all cfg 1000 dump1.all.*.cfg mass type xs ys zs fx fy fz id type c_eng
min_style cg
minimize 1e-25 1e-25 50000 100000
variable natoms equal “count(all)”
variable teng equal “c_eatoms”
variable length equal “lx”
variable ecoh equal “v_teng/v_natoms”
print “Total energy (eV) = {teng};"
print "Number of atoms = {natoms};”
print “Lattice constant (Angstroms) = {length};"
print "Cohesive energy (eV) = {ecoh};”
print “All done!”
I did similar calculations for different systems using the following potential files with 2 atoms system shown above. But with different spacing which was chosen by critically evaluating the position values used in the reference which was again found at the nist repository:
Ag_Zhou04.eam.alloy
FeNiCr.eam.alloy
Al99.eam.alloy
Unfortunately, by using the following EAM method formula and the values of the pair-potentials, charge density and embedding function from the reference, the values that I get for potential energy is different from LAMMPS:
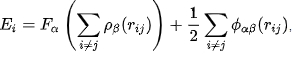
I have to format the EAM potential for the system which I am creating right now. Therefore, I want to make sure I understand how the LAMMPS algorithm is calculating the given values of potential energy.
I really appreciate your help. If you need any other information from my end, please, don’t hesitate to reach me out at: rjagat2@…2061….
Thanks.
Best,
Ritesh
Thanks.