Hello people!
I am trying to calculate the thermal conductivity of phosphorene. I used the Green kubo methode with a tersoff potential. I have a problem with the simulation code because I am not able to achieve the desired value.
if you have some remarks to improve my simulation I ll be thankful.
#################################################################################
units real
variable T equal 300
variable V equal vol
variable dt equal 4.0
variable p equal 200 # correlation length
variable s equal 10 # sample interval
variable d equal $p*$s # dump interval
so that the special characters do not get misformatted.
Also, we do not know what you mean when you say your results are wrong. What do you expect to see? How do you know you should expect that? And what do you see instead?
I meant that I have found results that differ from the values presented in the lectures.
It means that I got wrong result.
I you have remarks on my input, because I think something is wrong in there. just I want to know that the code simulation is ok and then I will look if I can modify the tersoff parameters. Thanks!
units real
variable T equal 300
variable V equal vol
variable dt equal 4.0
variable p equal 200 # correlation length
variable s equal 10 # sample interval
variable d equal $p*$s # dump interval
# convert from LAMMPS real units to SI
variable kB equal 1.3806504e-23 # [J/K] Boltzmann
variable kCal2J equal 4186.0/6.02214e23
variable A2m equal 1.0e-10
variable fs2s equal 1.0e-15
variable convert equal ${kCal2J}*${kCal2J}/${fs2s}/${A2m}
# setup problem
dimension 3
boundary p p p
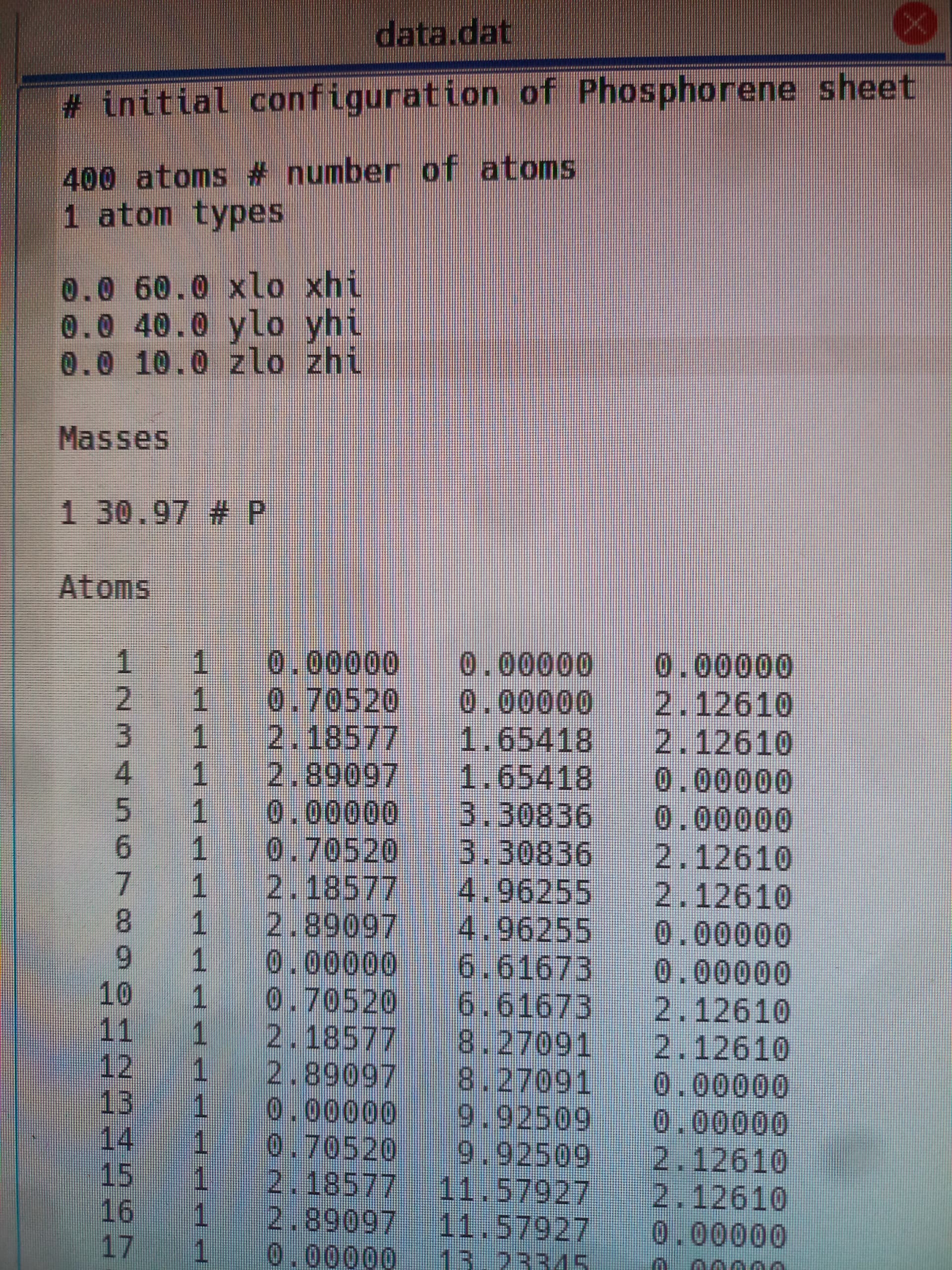
read_data data.dat
#region box block 0 24 0 16 0 3
#create_box 1 box
#create_atoms 1 box
pair_style tersoff
pair_coeff * * P.tersoff P
timestep ${dt}
thermo $d
# equilibration and thermalization
velocity all create $T 102486 mom yes rot yes dist gaussian
fix NVT all nvt temp $T $T 10 drag 0.2
#run 8000
run 1000
# thermal conductivity calculation, switch to NVE if desired
#unfix NVT
#fix NVE all nve
reset_timestep 0
compute myKE all ke/atom
compute myPE all pe/atom
compute myStress all stress/atom NULL virial
compute flux all heat/flux myKE myPE myStress
variable Jx equal c_flux[1]/vol
variable Jy equal c_flux[2]/vol
variable Jz equal c_flux[3]/vol
fix JJ all ave/correlate $s $p $d &
c_flux[1] c_flux[2] c_flux[3] type auto file J0Jt.dat ave running
variable scale equal ${convert}/${kB}/$T/$T/$V*$s*${dt}
variable k11 equal trap(f_JJ[3])*${scale}
variable k22 equal trap(f_JJ[4])*${scale}
variable k33 equal trap(f_JJ[5])*${scale}
dump dump_1 all custom 100 yt.dump id type x y z ix iy iz vx vy vz
thermo_style custom step temp v_Jx v_Jy v_Jz v_k11 v_k22 v_k33
run 100000
#variable k equal (v_k11+v_k22+v_k33)/3.0
variable k equal v_k11
variable o equal v_k22
variable ndens equal count(all)/vol
print "Average thermal conductivity armchair: $k[W/mK] @ $T K, ${ndens} /A\^3"
print "Average thermal conductivity ZigZag: $o[W/mK] @ $T K, ${ndens} /A\^3"
Did the lecture you referenced mentions how it gets the thermal conductivity values? There are a handful of factors that can affect the results of thermal conductivity. To list a few I can think of:
experimental factors (defects, impure samples, …)
the theoretical method (Green-Kubo vs. NEMD vs. BTE …)
the force field
the length of simulation for sampling heat flux, as well as the correlation length for ave/correlate (both should be long enough for convergence)
statistical errors (you may run the script with different random seed for initial velocities to check that)
NVE vs. NVT (remove comment for that “switch to NVE” part to check)
the method of integration (sometimes the correlation function fluctuates heavily and it’s tricky to do the integration correctly)
the size of system simulated (you may need a sufficiently large supercell to get the result converged)
method of calculating atomic stress (stress/atom vs. centroid/stress/atom; you probably have no choice here, but it can be a source of error since you have manybody forces)
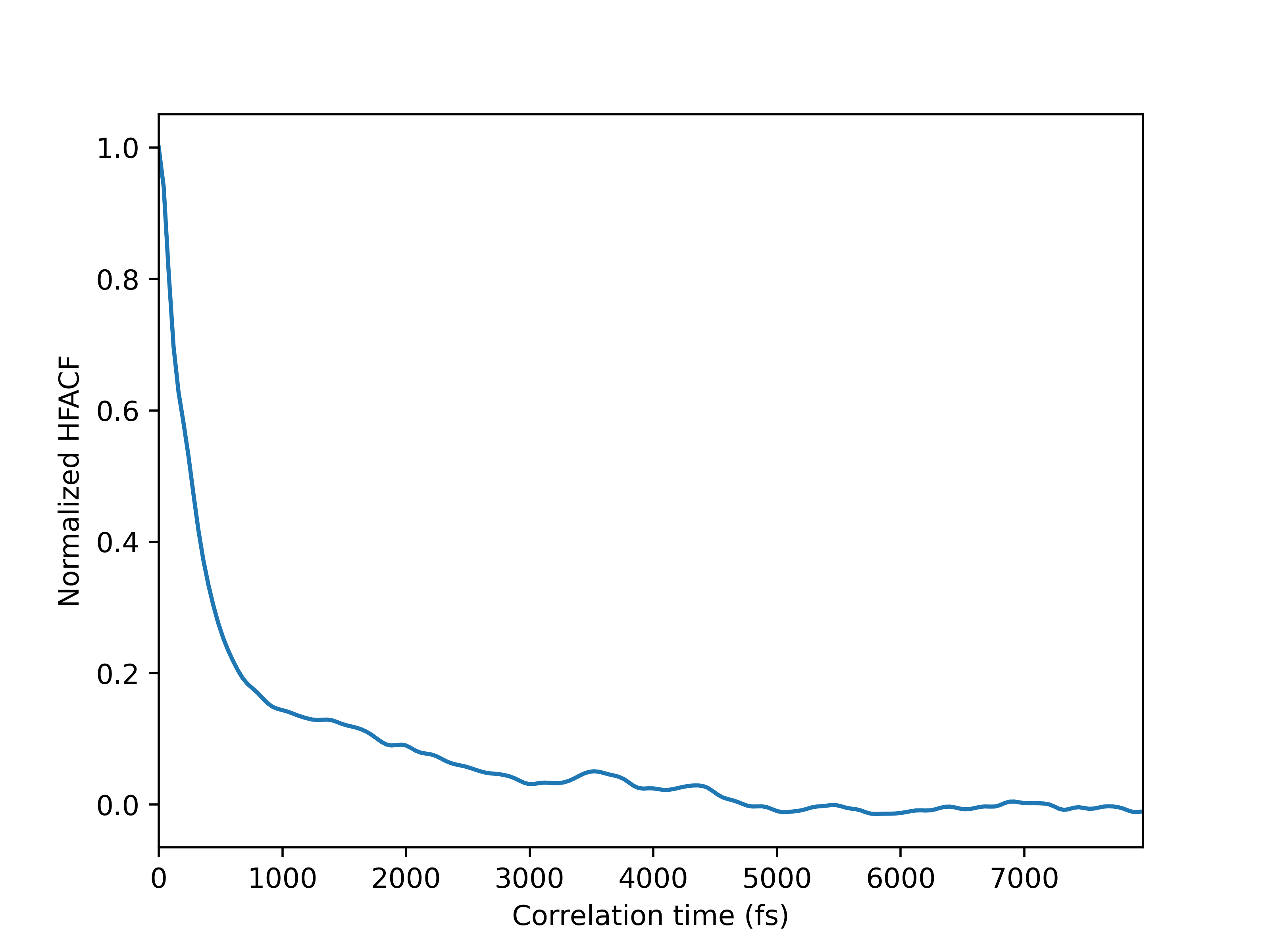
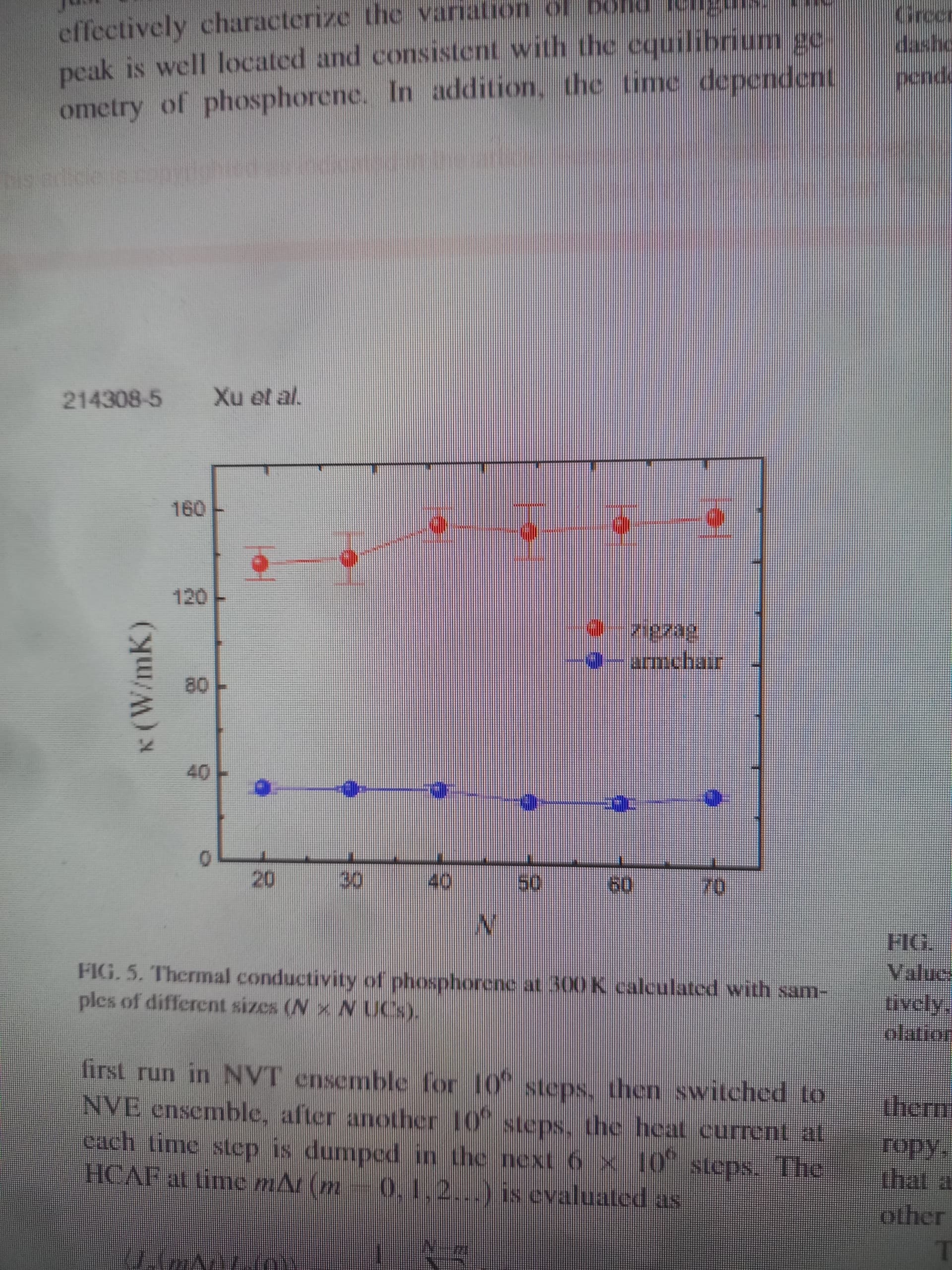
I took a (5-5) phosphorene sheet as a starting example. It is clear in the code that I used Green-kubo method with a tersoff potential (if someone has a reference of a useful tersoff parameters) . I got very small value of the thermal conductivity
the authors used NVE for calculating heat current, while yours use NVT;
the authors used much more steps for both equilibration and NVE runs than you;
the smallest cell in the figure is 20x20 UCs(=unit cell?).
btw: I believe you’re aware of this already, but just in case, make sure that you’re calculating in the same directions as the cited paper did. Sometimes external tools mess up the definition of axes when exporting lammps data files (I’ve personally encountered this).
the authors run both NVT and then NVE to calculate the heat current. I did the same I fixed to NVT for 10000 for equilibration and thermalization and then I fixed to NVE for 1000000 to calculate the heat current. But I still got a very weak value (~0.01…W/mk).
You worked with Lennard-jones potential, in my case I am trying to use tersoff potential to find the thermal conductivity of phosphorene. If the input file is fine then it should be something wrong with defining tersoff parameters for phosphorene?
With (1) such a small z-direction (2) periodicity in the z-direction, it is very likely that your sheet is interacting with z-periodic copies of itself. I would recommend trying a z-height of 60 angstroms.
We increased the size of the sheet to (20-20) UCs (1600 atoms) also as you recommend we adjusted the z-height to 60 angstroms. The final result is still not good (0.20 W/mk, 0.23 W/mk for armmchair and zigzag resperctively)
Hello brother.
Did you solve your problem about the result?
What was your problem in the code, Can you shear with me?
Actually I am also facing the same problem.