Dear Lammps users,
I encountered a stress calculation problem when simulating three layers of hexagonal boron nitride (hBN) sliding over three layers of graphene (Gr) under normal loads. In my simulation system, the hBN layers are named tip1l, tip2l and tip3l, and the Gr layers are named sub1l, sub2l, sub3l, from top to bottom. To fix the the top hBN layer, i.e. tip1l, as rigid while allow it free to move in the z direction, I used the following command
fix rig tip1l rigid single force * off off on torque * off off off
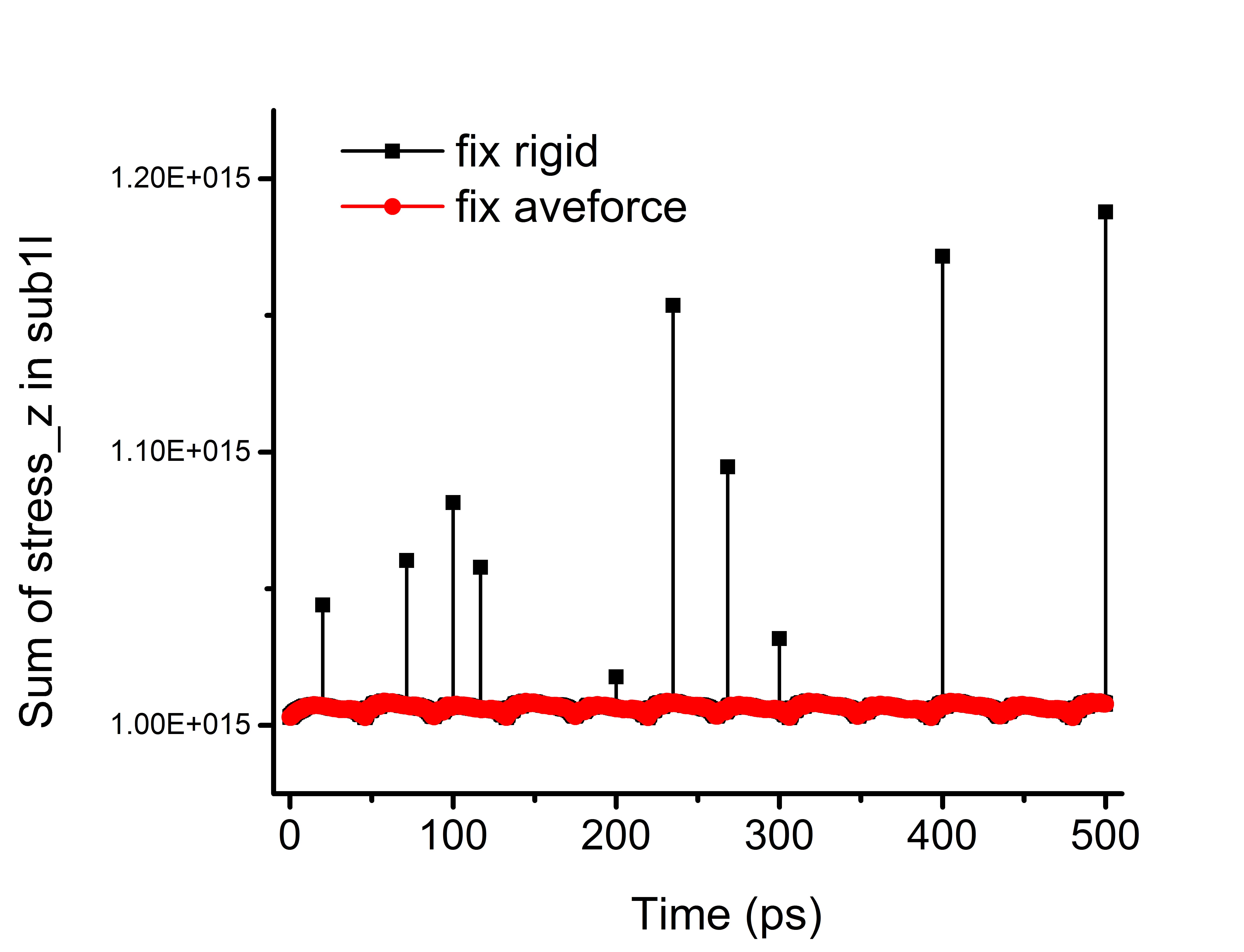
In the x and y directions, constant sliding velocities are set by velocity command. The bottom layer, i.e. sub3l is also fixed rigid by not updating the atomic positions. I output the atomic stresses in the flexible layers, e.g. in the top Gr layer (sub1l). I find at some time steps there are atoms with stresses in the z direction much larger than nearby atoms and those atoms are located near the CPU grid lines. Please see the attached figure 1 where 16 cores are divided into 441 grids. The blue and red colored atoms are the abnormal atoms. Then, I calculate the sum of the square of the atomic stresses (z direction) in the layer and output it every 10 time steps. I find abnormal spikes (corresponding to the occurrence of abnormal stresses) occurred at the timesteps (multiples of 100 ps) I output restart files and also at some non-restart-output timesteps (see attached figure 2). I think those timesteps are the ones LAMMPS rebuild the neighbor list.
Then I changed the fix rigid with fix aveforce to set the top layer as rigid in an alternative way. (The atomic masses for B and N are unified to their average in the tip1l). The abnormal spikes are gone. Please see attached figure 2.
So now, I think the abnormal stress is related to fix rigid at time steps that the neighbor list is rebuilt. The atoms close to the CPU grid lines may migrate from one CPU to another CPU. In such case, the force (in the direction that is turned on) calculation may not be done correctly. Both LJ and ilp/graphene/hbn potentials are tested. They present similar behavior.
I would like to know your thoughts on this issue. The Lammps version is 24 Aug 2020. Please contact me for typical data file and input file as they are too large to attach.
Thanks and best,
Xiang Gao
I’m not following the details of your explanation, but I’ll note that the fix rigid command has a virial yes/no option:
The fix_modify virial option is supported by these fixes to add the contribution due to the added forces on atoms to both the global pressure and per-atom stress of the system via the compute pressure and compute stress/atom commands. The former can be accessed by thermodynamic output. The default setting for this fix is fix_modify virial yes.
So that may be affecting what you are seeing in your stress per layer output. Note the rigid constraint for a body implies additional virial terms, so defining or summing up per-atom stress for a rigid body is non-trivial.
Steve
Hi Steve,
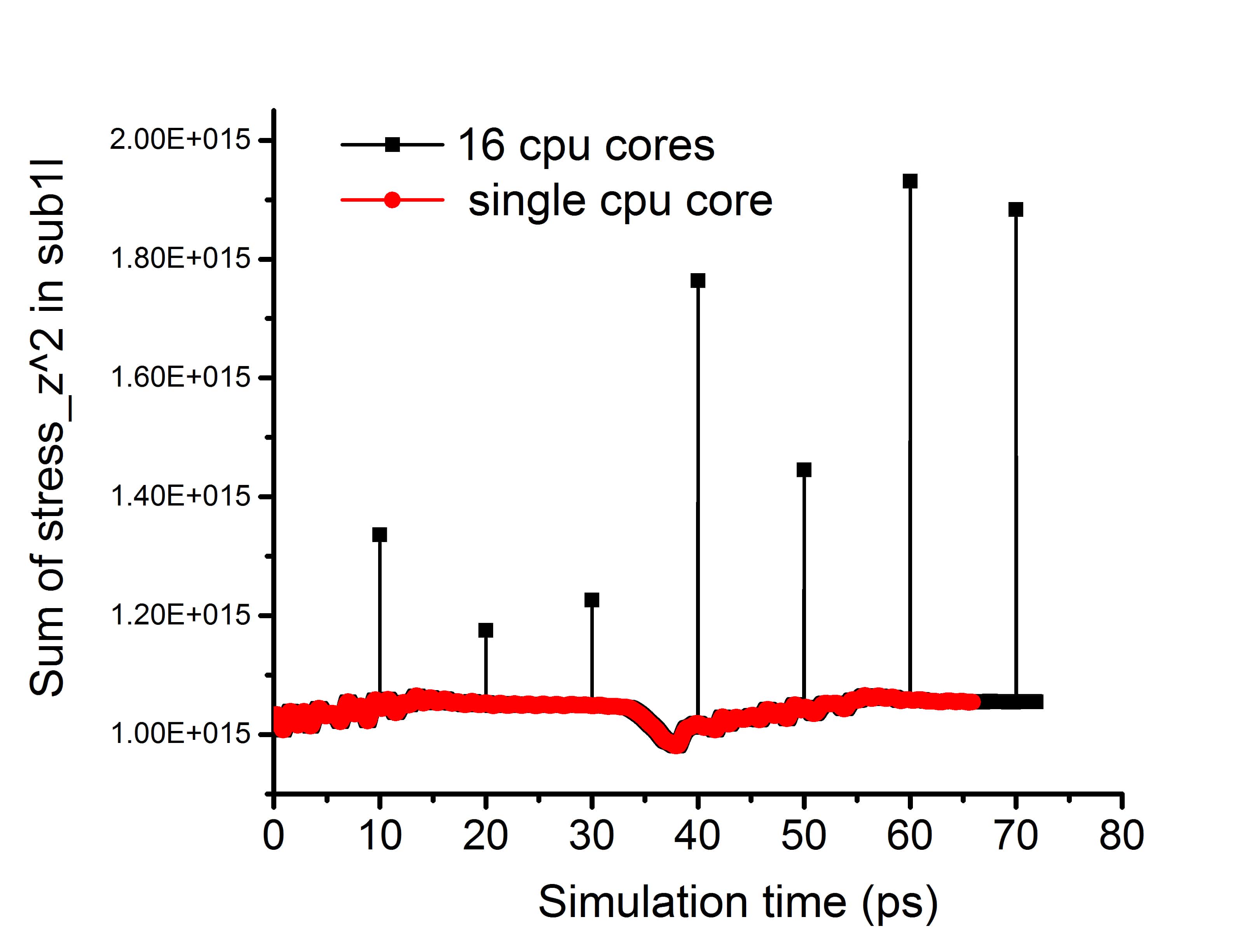
Thanks for your reply. Sorry for not explaining my problem clearly. I am not considering the atomic stress on the rigid layer, but the flexible layers (without fix rigid) underneath it. Once I use fix rigid on the top layer, I found abnormal atomic stresses on the flexible layers (probably every time) when the neighbor list is rebuilt. This problem only occurs with parallel computation, while single core computation not. Please see attached figure. The abnormal stressed atoms are only located at the cpu subdomain grid lines. I guess there may be some communication issues in such case. And this issue is probably related to fix rigid, as the abnormal stress is gone when replacing fix rigid with fix aveforce.
Best,
Xiang
Hi Steve and Lammps users,
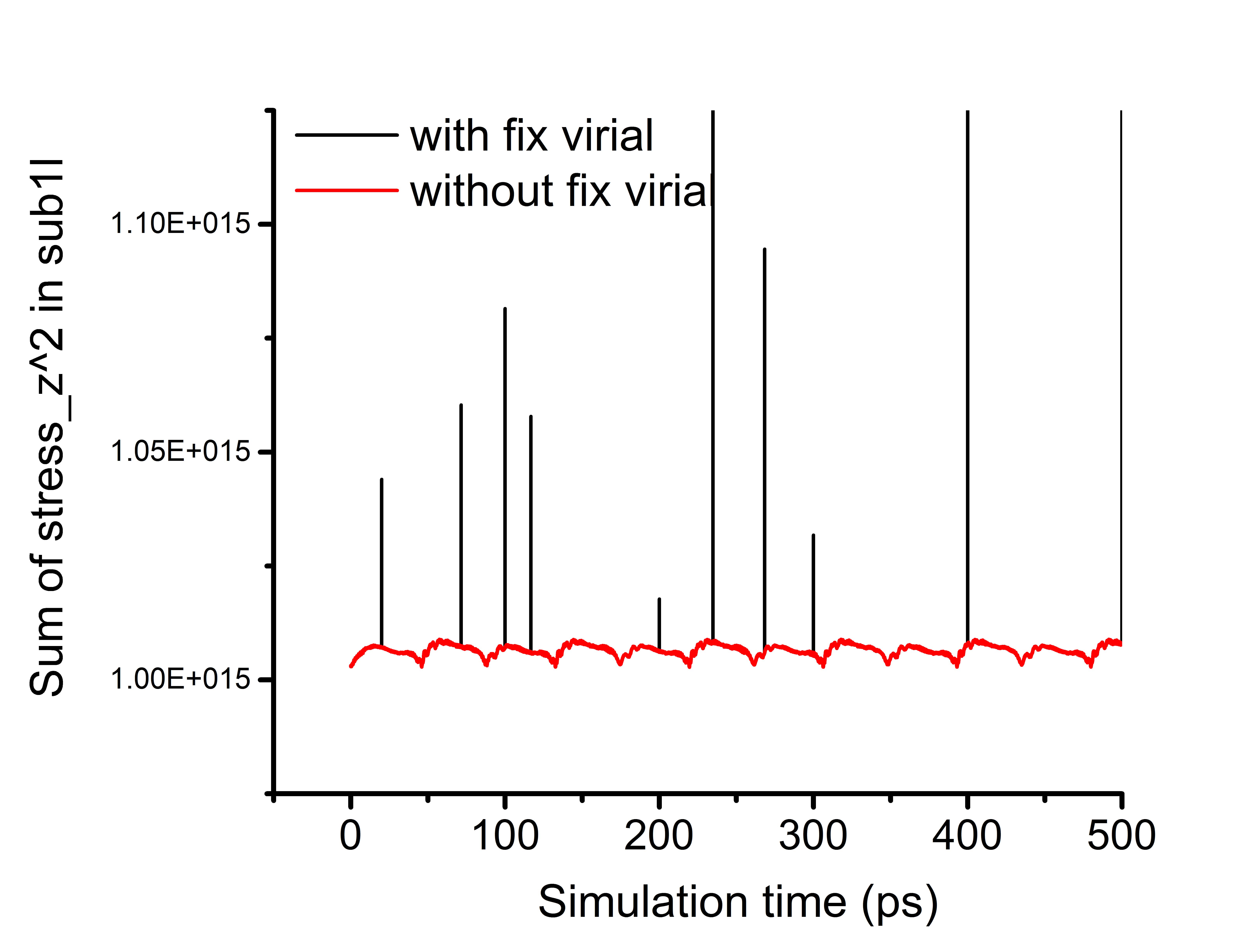
Here is an update and a short summary with my issue. I applied fix rigid to a group of atoms, then I found weird atomic stresses on atoms not belonging to the fix rigid group. By setting the fix_modify virial no, the weird atomic stresses are gone (see attached figure). The weird stress behavior seems only to occur in parallel computations at the time steps when lammps update the neighbor list. The atoms with weird stress are very close to the CPU subdomain grid lines, indicating this issue may be related to the migration of atoms. I am confused how the virial term due to fix rigid affects the stresses of atoms not in the fix rigid group. I will appreciate it very much if anyone has some thoughts on this.
Thanks and best,
Xiang
I have set up a small test case (included below in case you are curious) for this and running this with valgrind in parallel shows that there is uninitialized data access. this requires some debugging effort to correctly indetify the source of this and implement a solution. more on this later.
axel.
lattice fcc 1.0
region box block -2 2 -2 2 -2 2
create_box 2 box
create_atoms 1 box
region layer block EDGE EDGE -0.5 0.5 EDGE EDGE
group layer region layer
set group layer type 2
mass * 1.0
pair_style lj/cut 2.0
pair_coeff * * 0.0 1.0
pair_coeff 2 2 1.0 1.0
group bulk subtract all layer
fix 1 bulk nve
fix 2 layer rigid single
neigh_modify every 2 delay 0 check no
compute 1 all stress/atom NULL virial
dump 1 all custom 1 dump.lammpstrj id type c_1[*]
Hi Axel,
I followed and slightly modified your script. I find the problem can be reproduced. Please see attached figure. There are few very large spikes when using parallel computation. I guess at those time steps the neighbor list is rebuilt.
lattice fcc 1.0
region box block -2 2 -2 2 -2 2
create_box 2 box
create_atoms 1 box
region layer block EDGE EDGE -0.5 0.5 EDGE EDGE
group layer region layer
set group layer type 2
mass * 1.0
pair_style lj/cut 2.0
pair_coeff * * 0.0 1.0
pair_coeff 1 2 1.0 1.0
pair_coeff 2 2 1.0 1.0
group bulk subtract all layer
fix 1 bulk nve
fix 2 layer rigid single force * off off on torque * off off off
neigh_modify every 2 delay 0 check no
compute 1 bulk stress/atom NULL virial
variable 1 atom c_1[3]^2
compute 2 all reduce sum v_1
thermo 100
thermo_style custom step temp ke pe etotal c_2
thermo_modify flush yes format float %15.5g
#dump 1 bulk custom 1 dump.lammpstrj id type x y z c_1[*]
restart 10000 restart.1 restart.2
run 2000000
Hi Axel and Steve,
May I follow up if you have any new comments on this stress issue? In brief, the fix rigid command affects the atomic stresses on the atoms without the fix rigid constraint in parallel computation via virial term. I understand this issue can be avoided by using fix_modify virial no. But I hope to understand what is the reason for this problem with your help.
Thanks very much!
Best,
Xiang