Dear all,
The DPD method implemented in LAMMPS is the one reported by Groot and Warren in 1997,
and there are some new modifications of the original dpd method, specifically “many-body DPD”.
One of the most important MDPD paper can be Phys Rev E, 68, 066702 (2003) by P. B. Warren,
who modify the force in the conservative part by including a density-dependent force,
which enables the simulation of liquid-vapor interface.
I am not good at cpp and have failed to modify the source code myself.
I would like to know whether there is someone have done this job before,
or I would expect the package maintainer to help me to implement it?
Looking forward to any kind suggestions~
Chen Chen
Dear all,
dear chen chen,
I am not good at cpp and have failed to modify the source code myself.
I would like to know whether there is someone have done this job
before,
or I would expect the package maintainer to help me to implement it?
why would you "expect" the package maintainer to help you? this is
the 'cruel' world of open source software. if you want it done, you
either have to do it yourself (LAMMPS is about the easiest code to
extend with a modified pair potential that i've run across so far),
convince somebody to do it for you (make a more compelling argument)
or pay somebody to do it (hire an intern that can program).
cheers,
axel.
Hi Axel,
Thank you for your explanation about the cruel world, hoho~
I would try once more~
Chen Chen
Dear all,
As I mentioned a week ago, I am trying to implement PairMDPD by choose PairDPD as a template.
I tried to modify the PairDPD:compute according to the paper of Warren.
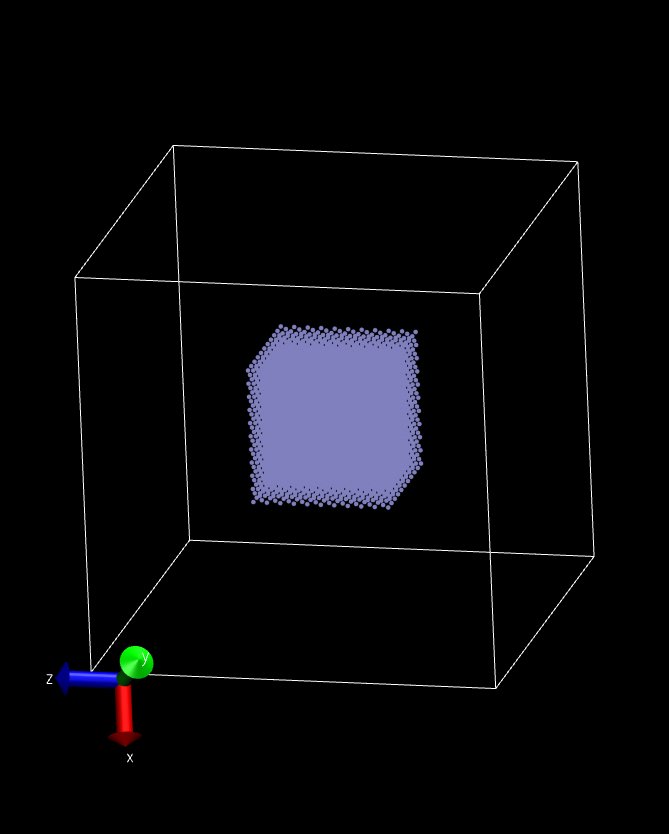
If implemented was successful, I would get a drop in the box derived from a fcc cube (Fig. 1)!
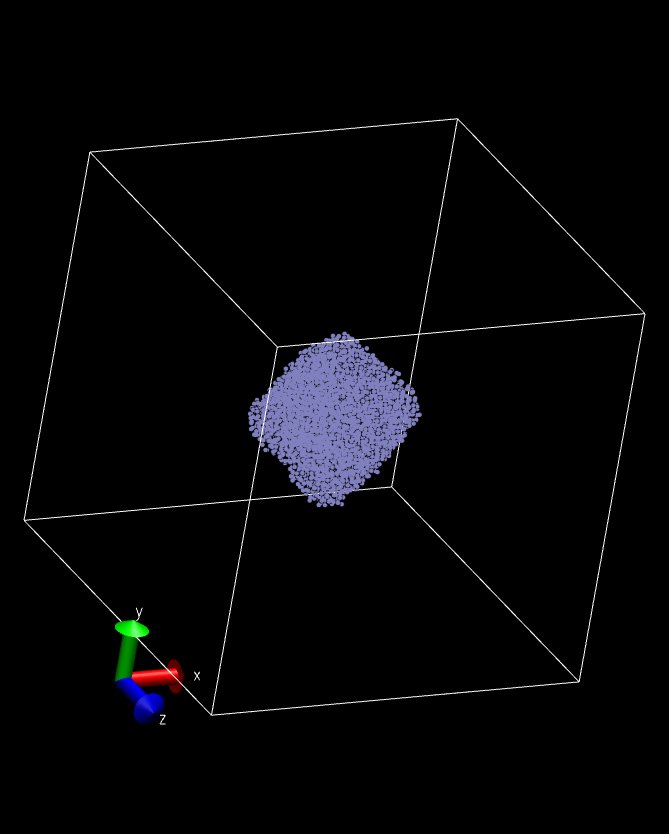
It is encouraging that I can get a spherical drop on only one processor! (Fig. 2)
But get an octahedron on 8 processors (2x2x2) (Fig. 3)
I know something is missed and thus I did not get multi-processor to work appropriately.
But I could not figure out where to modify, and it really bothered me a lot…
Could anyone give some hints?
Attached are the modified files: pair_mdpd.h, pair_mdpd.cpp, style_user.h and 3 snapshots~
pair_mdpd.cpp (11.7 KB)
pair_mdpd.h (1.3 KB)
style_user.h (1.63 KB)

I’ve forgotten the input file:
No one is likely to debug your code for you. I'd suggest
printing thermo output every step. If you don't get
good agreement every step on 1 versus more procs,
at least for the first few steps, then you should be able
to figure out why. If MDPD uses random #'s you'll
likely need to turn that term off in the force field.
Steve