Dear Aidan,
As you may remember, I'm attermpting to carry out simulations of
crystalline ZrO2 using the reaxFF potential. I've been unable to
equilibrate the system at finite temperature, so I checked the
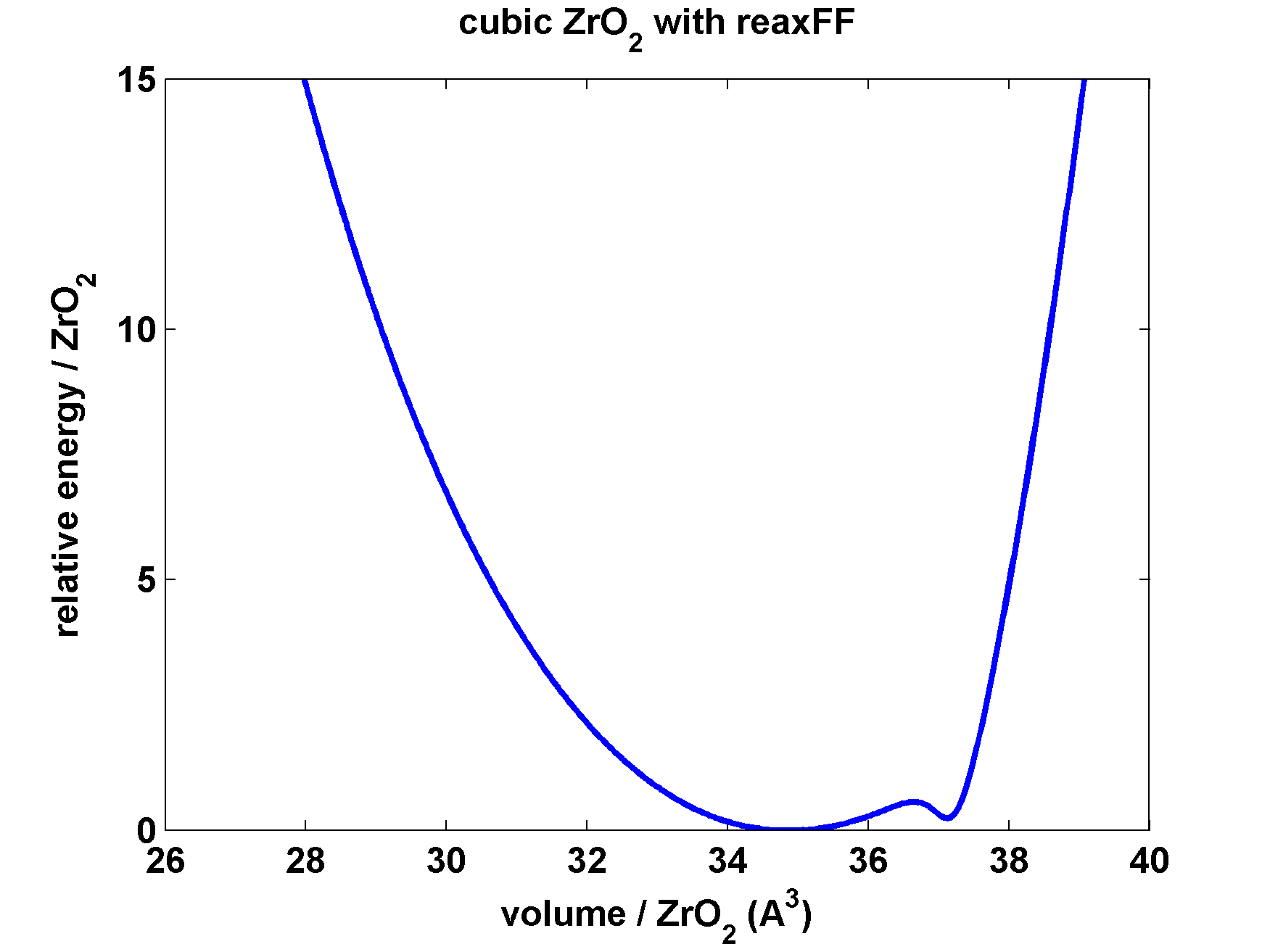
zero-T energy per atom as a function of lattice parameter, which
gives a measure of the interatomic potential in the crystalline
environment. This turns out to have a double minimum (plot column
14 as a function of column 13), which is unphysical. I can think
of several possible causes of this error:
(1) My LAMMPS script (below) contains an error. It seems unlikely,
because it's very simple, but could you please have a look at it?
(2) The results aren't coverged. However, I've checked that they're
independent of the parameters governing hbcut and the charge
equilibration precision, and the double minimum is robust to
variations of the lower and upper limits to fix deform.
(3) The potential I'm using (published as Supplementary Information
to J. Chem Phys 112, 3133 (2008), and attached) contains an error. I've written
to the authors but haven't yet received a reply.
(4) LAMMPS and the reaxFF libraries weren't properly compiled /
linked on my system. Could a problem here cause the observed
behaviour? If so, would it be possible for you to run my script on
your system and compare the results?
Thanks for your help,
Philip
############### Parameters ########################################
######## parameters for the potential ############################
# cutoff distance for hydrogen bonds in Armstrongs
variable hbcut equal 1
# relative accuracy of precision for charge equilibration
variable charge_precision equal 1.0e-6
######## parameters for structure ################################
# number of unit cells in x-, y- and z-directions:
variable size equal 6
# initial lattice constants:
variable ax_i equal 4.8
variable ay_i equal 4.8
variable az_i equal 4.8
# final lattice constants:
variable a_f equal 5.4
# number of steps for deformation:
variable deform_steps equal 41
# final size of simulation cell
variable L_f equal \{a\_f\}\*{size}
############## Initialize ########################################
boundary p p p
units real
atom_style charge
variable relca equal \{az\_i\}/{ax_i}
variable relba equal \{ay\_i\}/{ax_i}
lattice custom \{ax\_i\} a1 1\.0 0\.0 0\.0 a2 0\.0 {relba} 0.0 a3 0.0 0.0 ${relca}&
origin 0.0 0.0 0.0 basis 0.0 0.0 0.0 basis 0.5 0.5 0.0 basis 0.5 0.0 0.5 basis 0.0 0.5 0.5&
basis 0.25 0.25 0.25 basis 0.75 0.25 0.25 basis 0.25 0.75 0.25 basis 0.75 0.75 0.25 basis 0.25 0.25 0.75 basis 0.75 0.25 0.75 basis 0.25 0.75 0.75 basis 0.75 0.75 0.75
region mybox block 0 \{size\} 0 {size} 0 ${size}
create_box 2 mybox
create_atoms 2 box basis 1 2 basis 2 2 basis 3 2 basis 4 2&
basis 5 1 basis 6 1 basis 7 1 basis 8 1&
basis 9 1 basis 10 1 basis 11 1 basis 12 1
# atom 1 is Oxygen, atom 2 is Zr
group O type 1
group Zr type 2
set group O charge -2
set group Zr charge 4
mass 1 16
mass 2 91.22
neigh_modify one 20000 page 200000
############### Definition of potential #########################
pair_style reax \{hbcut\} {charge_precision}
pair_coeff * * ffield.reax 3 5
log Murnaghan_hb${hbcut}_p${charge_precision}.dat
############### Deformation of unit cell #######################
variable ax equal lx/v_size
variable ay equal ly/v_size
variable az equal lz/v_size
variable vol_per_ZrO2 equal vol/(\{size\}\*{size}*\{size\}\*4\)
\# Shift the origin of the energy scale to \(approximately\) the
\# energy of the ground\-state monoclinic structure
variable E\_per\_ZrO2 equal etotal/\({size}*\{size\}\*{size}*4)+565.76
thermo_style custom step press pxx pyy pzz etotal pe evdwl ecoul v_ax v_ay v_az v_E_per_ZrO2 v_vol_per_ZrO2
thermo_modify flush yes
thermo 1
fix DEF all deform 1 x final 0 \{L\_f\} y final 0 {L_f} z final 0 \{L\_f\} units box
run {deform_steps}
unfix DEF
ffield.reax (17.5 KB)