Dear All,
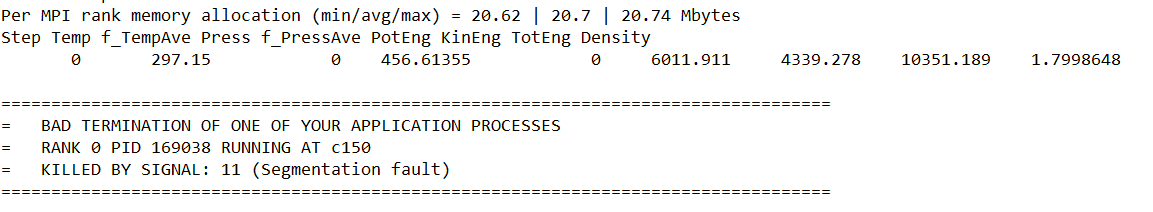
I am simulating 50 molecules (approx 4000 atoms) in a box with periodic boundary condition. I simulate in the order NPT, NVT and finally NVE. In the NVE run, I want to add a CO2 molecule to the simulation. I am encountering the error in the NVE simulation ‘Bond atoms missing on procs’ (image below).
I would just like to add a brief on my simulation for clarity (the major steps).
read_data ./data.system group all extra/atom/types 2 extra/bond/types 1 extra/angle/types 1 extra/bond/per/atom 2 extra/angle/per/atom 1 # read the positions
mass 8 12.010700
mass 9 15.999400
include forcefield.data # read the force field
molecule CO2 ./co2lammps.txt boff 15 aoff 34 # read co2 molecules
Using read_data I fetch data of all the atom (other than CO2). I create some space for new bonds and angles using ‘extra’. Forcefield.data includes all the parameters including the one’s I would be using for CO2. The data for CO2 is stored on the ‘molecule’ template. I do not use ‘offset’ in the molecule template as I offset the atom type later using ‘fix deposit’
In the NVE run, I include the following commands to load the CO2 molecule which is of the rigid type
region myblock block {xlow} {xhig} {ylow} {yhig} {zlow} {zhig}
fix constrain COtwo rigid/small molecule mol CO2
fix co2 COtwo deposit 1 7 1000 10000 region myblock mol CO2 rigid constrain units box
I specify the type as 7 as I would like to have type 8 and 9 for my new atoms. I create the region as my entire simulation box. I’ve created a group-id ‘COtwo’ earlier in the simulation file which consists of ‘C’(8) and ‘O’(9) atoms of CO2.
Besides, if I add ‘near 0.1 attempt 10000’, I still get the same error. If I add ‘near 2’ instead, the simulation is killed (image below)
I seek your input on this issue. What am I doing wrong while inserting a new molecule into the box? For reference, I am also attaching my simulation and CO2 molecule file in case required. If any further information is required, please do let me know.
Thank you very much.
Regards,
Kunal Mavani
co2lammps.txt (399 Bytes)
Simulation.in (5.6 KB)
depositing molecules is more tricky than depositing atoms. you have to be very careful to avoid close contacts of atoms.
since the problems happens immediately, you need to figure out what is exactly happening, best by dumping coordinates, velocities, and forces in every step and checking out whether there are any unwanted overlaps causing atoms to experience extreme forces and thus leading to the error you observe.
axel.
Dear Axel,
Thank you for your response. I note the coordinates at every timestep. I see that a new molecule is deposited at timestep 1, whereas I specify the value of M(deposit the molecule every timestep) as 1000. I start the NVE run by resetting the time to 0. Why does the deposition take place at timestep 1 instead of 1000?
The LAMMPS version I use is 22Aug2018.
Regards,
Kunal Mavani
fix deposit does insert every M steps. if you ask for every 1000 steps you will get insertions on steps 1, 1001, 2001, 3001 and so on.
there are multiple reasons not to do it on steps 0, 1000, 2000, 3000 and so on.
if you want to avoid close contacts, you have to do one of three things:
- make sure the region where you insert new atoms does not overlap with the region where atoms currently are (note the lengths may differ if you are not using either units box or units lattice consistently).
- make sure that deposited atoms can move away from where they get deposited in time before another deposit could happen.
- avoid depositing atoms close to existing atoms (see keyword near). please note that when inserting molecules, you have to take into account that the near keyword considers the “insertion center”, not the individual atoms of the inserted molecule, so you may need to increase value to take the extent of the inserted molecule into account.
axel.
In a new simulation in which I noted coordinates at every timestep, the simulation ran till timestep 4. I note distances in the table below. The VDW for each atom is about 3-3.5 A. the distance is clearly less than that in timestep 1. The repulsive forces are infinite I guess then. In addition to my previous mail why the deposition is taking place at timestep 1 instead of 1000, is there a way to somehow rectify this?
Timestep New added atom Atom kicked out
1453 1463
1 4901 2.226054 2.666325
4902 3.166573 3.648739
4903 1.604749 1.895902
2 4901 11.59446 10.14456
4902 12.64877 11.16772
4903 10.56157 9.153234
3 4901 22.25896 23.82405
4902 22.90494 24.4623
4903 21.65628 23.22664