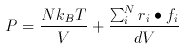Dear Steve and all other lammps users,
when I set the boundary style to be "f " , I output the thermo_press[3], but some "lost atoms" error occurs.
after the boundary style was set to be "s", there was no error. but the thermo_press I get is something different with that when boundary style is "f"
in lammps manual, the thermo_press is defined to be
I am wondering when the boundary style has been changed, is there volume change during the simulation. if not , why the thermo_press[3] are different for two boundary style.
my output script is :
fix 2 all ave/time 2 20 100 c_thermo_press[3] file stress.all
boundary style is
boundary f f f
or
boundary s s s
thank you
L i Ming