Dear All,
I am new to modeling Hydrocarbons and using Reaxc to model chemical bonding. I used an online tool to create a chain, and using these atom coordinates; I wanted to simulate the chain and get its density and check the force-field with this measurement. Unfortunately, I couldn’t use the NPT ensemble as the simulation breaks after a couple of thousands of time steps. I have noticed in the literature, and in the LAMMPS examples the codes are using NVT or NVE, but those fixes will not modify the volume resulting in a correct density.
I would be grateful to know the correct approach to get the right density using the ReaxFF potential style.
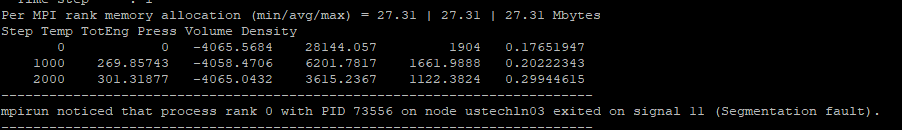
The output of the LAMMPS code using the NPT ensemble on one chain of dodecanethiol.
Amir Behbahanian
Graduate Research and Teaching Assistant
Mechanical & Aerospace Engineering
Utah State University
4110 Old Main Hill
Logan, Utah 84322
[email protected]
ReaxFF is an “expensive” potential which computes a lot of properties based on the environment of atoms, which is stored. This storage is estimated at the beginning of a run, but because of that, the pair style doesn’t handle the situation well, where you have large changes in the environment, e.g. a significantly increasing number of neighboring atoms.
Starting such a system and so far away from equilibrium is extremely wasteful and inefficient.
A better way would be to a) try to create a more “packed” system from the beginning (check out tools like packmol) and b) pre-equilibrate the system using a regular classical force field, e.g. OPLS/AA. Once you have generated a system at the desired density you can strip off all bonded interactions (and remove the bonds, angles, dihedrals without deleting the atoms) and then continue with pair style reax/c.
Furthermore, I would follow a more direct approach to obtaining the desired density by using fix nve and fix langevin (with a somewhat short time constant to kinetic energy is dissipated fast) and then “compress” the system to within 10-20% of the target density by simply doing a run with fix deform. once that system has reached the target volume and temperature stabilized at the volume, you can switch to fix npt and finalize pre-equilibration. since this doesn’t have to be very accurate, you can use pair style lj/cut/coul/cut without kspace and thus run a pretty fast simulation. From there you can then switch to a ReaxFF force field much more easily and won’t have to deal with large changes to the environment.
Axel.