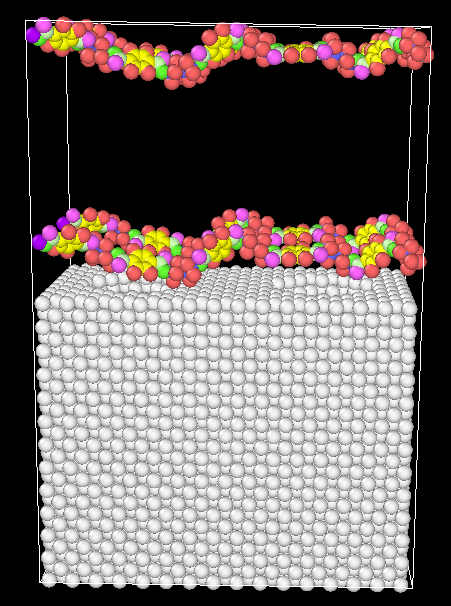
I try to investigate the adhesion property between metal and polymer, such as polyethylene and copper. So, I built a copper substrate and added some polymers on it and then combined them into one model by reading data twice. The polymer chain is long and have 500 repeated units. It is not easy to obtain the desired high density by Packmol or Materials Studio before being introduced into LAMMPS. When I used “fix deform” command, I found the metal substrate was simultaneously compressed and I think it is not right. Is there other commands or functions can help rationally compressing polymers into desired density without affecting the substrate? The model was attached
The version of LAMMPS I use is LAMMPS-8Apr21.
I would be really appreciated for any advices you provide.
Fix deform is meant to be used with periodic systems, but you should have a fixed non-periodic boundary in the direction of the slab and surface.
To avoid deforming/compressing the metal slab with fix deform, you could use the “dilate” keyword and define a group that contains only the polymer and then using that as argument to dilate. that will only compress the polymer atoms. in conjunction with fix deform, you need to also an integrator and thus the compression by fix deform to the metal atoms should not make much of a difference, since the metal can relax. it would only make a different if you would also immobilize the metal.
That said, because of the preference of non-periodic boundaries, you would need an alternate way, anyway. there are multiple possible options:
you can assign a velocity toward the surface to all polymer atoms with “velocity add”. they will then move toward the surface and once the atoms are closer they will attract each other
similarly you can use fix addforce to add a force to the polymer atoms to drag them toward the surface
you can use fix wall/harmonic with a variable wall position and move that wall during a run toward the surface
at any rate, it will be unphysical to enforce a specific density, since you have an open system where there is no confinement and thus the polymer will relax to the density it “wants” to be in. on top of that, to compute density you need the volume, but the volume of a subsystem in an atomic scale simulation is not a well defined entity.