Dear lammps-users,
I am trying to calculate contact angle of water droplet on K-illite surface, which is triclinic system. Figures of systems are presented below. I want to get the contour map of mass density for water. So, I used command “compute densitymap killite chunk/atom bin/cylinder z 0 0.5 56.6814301530825 70.8451929595014 0 0.5 70 nchunk every limit 0 ids every compress no discard mixed units reduced bound z 0 100” ,“fix waterdensity killite ave/chunk 10 10 100 densitymap density/mass norm sample ave running file densitymap.txt overwrite” to compute and output the density distribution. But, I got a wrong result “densitymap.txt”, in which the value of mass density for water is zero. I have checked my input file carefully for several times, but I could not find any problems in input script. My lammps version is 29-Oct-2020, lmp_intel_cpu_intelmpi, and GPU was used to acceleration. The input file was attached. Hope you give me some useful and valuable suggestions. Thanks.
Best Regards.
Dongbo Wang
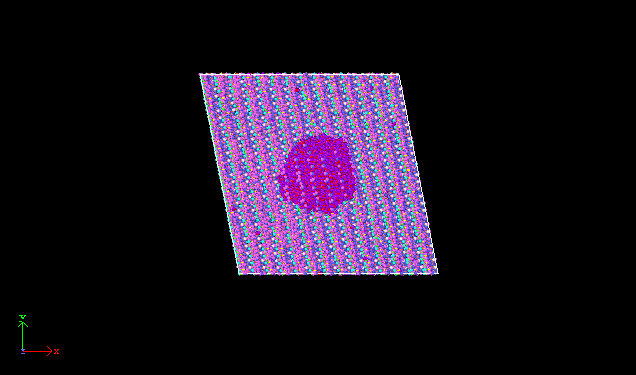
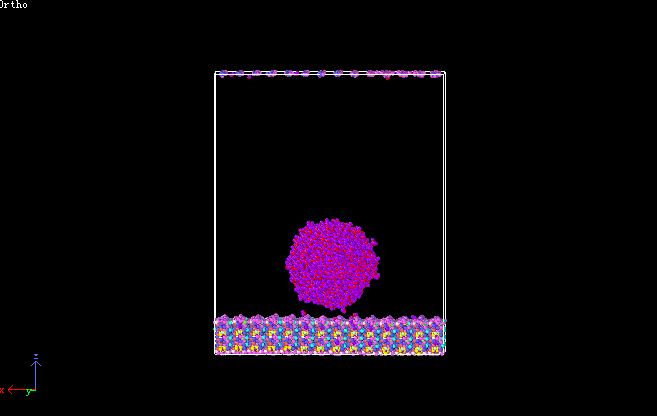
in.3 (2.42 KB)